

Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome
- PMID: 10655479
- PMCID: PMC15511
- DOI: 10.1073/pnas.97.3.1032
Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome
Abstract
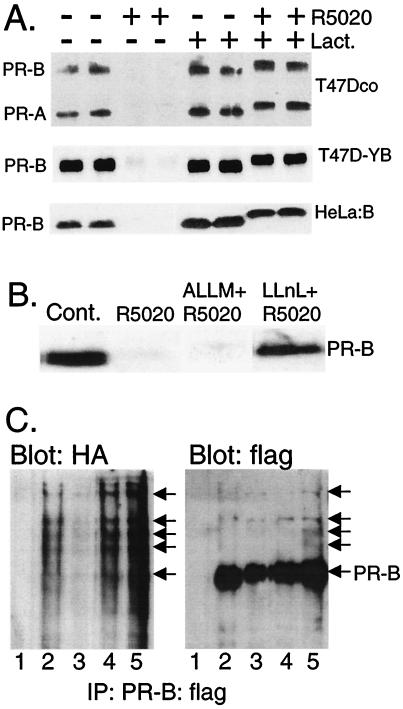
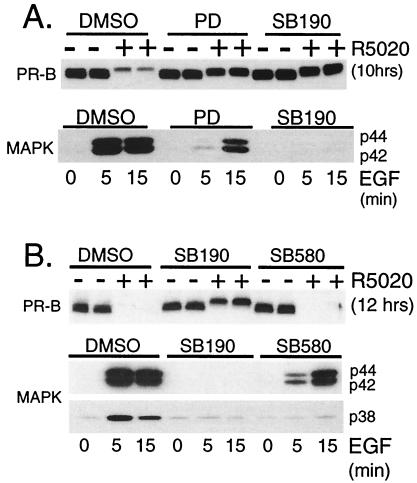
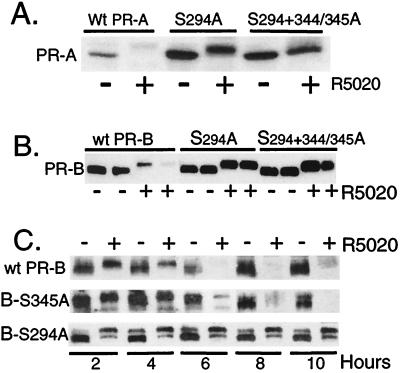
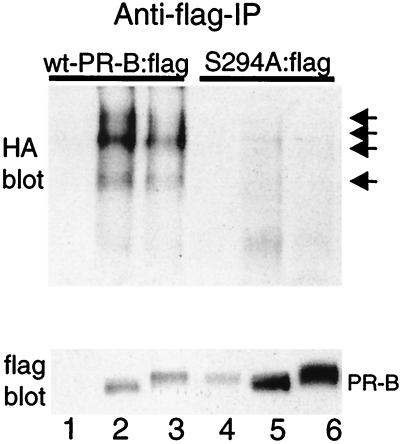
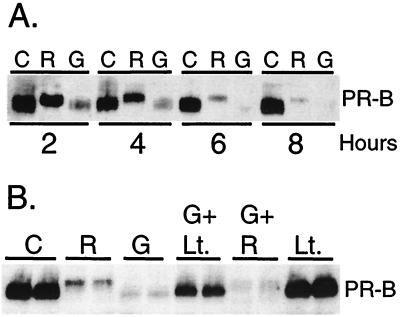
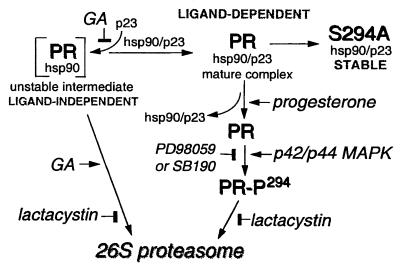
Ligand-dependent down-regulation that leads to rapid and extensive loss of protein is characteristic of several nuclear steroid receptors, including human progesterone receptors (PRs). In breast cancer cells, >95% of PRs are degraded 6 h after the start of progestin treatment. The mechanism for down-regulation is unknown. We examined the role of PR phosphorylation by mitogen-activated protein kinases (MAPKs) in this process. Lactacystin and calpain inhibitor I, specific inhibitors of the 26S proteasome, blocked progestin-induced down-regulation, and ubiquitinated conjugates of PR accumulated in cells. Ligand-dependent PR degradation was also blocked by specific inhibition of p42 and p44 MAPKs. To define the targets of phosphorylation by this kinase, two serine/proline MAPK consensus sites on PR were mutated. We demonstrate that mutation of PR serine-294 to alanine (S294A) specifically and completely prevents ligand-dependent receptor down-regulation. We also find that rapid, ligand-independent degradation of immature PR intermediates occurs by a proteasome-mediated pathway. These results demonstrate that PR destruction, by either of two alternate routes, is mediated by the 26S proteasome. Specifically, down-regulation of mature PRs occurs by a mechanism in which ligand binding activates PR phosphorylation by MAPKs at a unique serine residue, which then targets the receptors for degradation.
Figures
References
-
- Horwitz K B, McGuire W L. Steroids. 1975;25:497–505. - PubMed
-
- Elledge R M, McGuire W L, Osborne C K. Semin Oncol. 1992;19:244–253. - PubMed
-
- Wei L L, Krett N L, Francis M D, Gordon D F, Wood W M, O'Malley B W, Horwitz K B. Mol Endocrinol. 1988;2:62–72. - PubMed
-
- Nardulli A M, Katzenellenbogen B S. Endocrinology. 1988;122:1532–1540. - PubMed
-
- Mimnaugh E G, Chavany C, Neckers L. J Biol Chem. 1996;271:22796–22801. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
