

Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy
- PMID: 10655507
- PMCID: PMC15566
- DOI: 10.1073/pnas.97.3.1196
Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy
Abstract
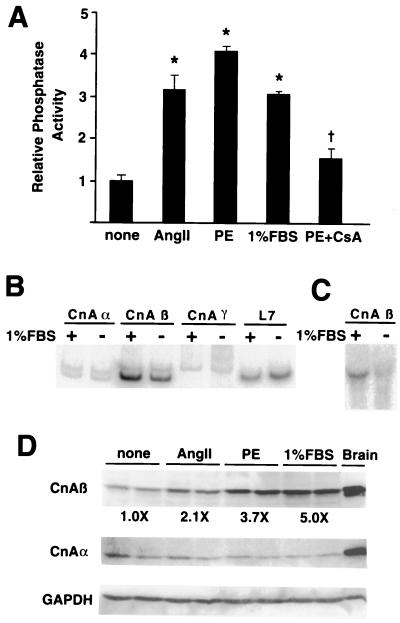
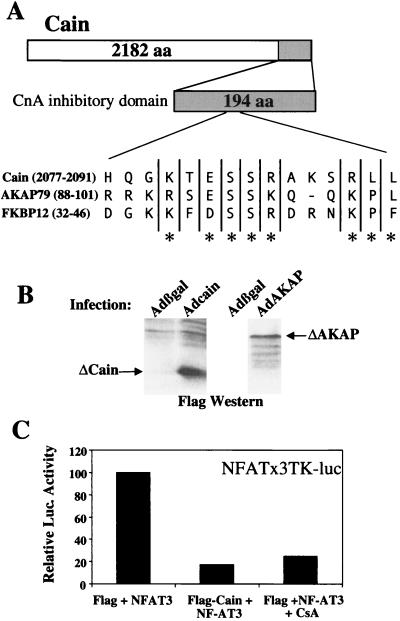
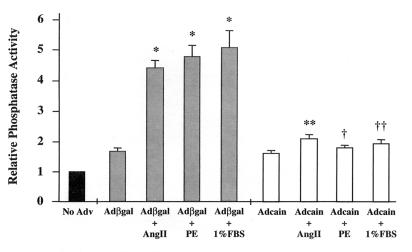
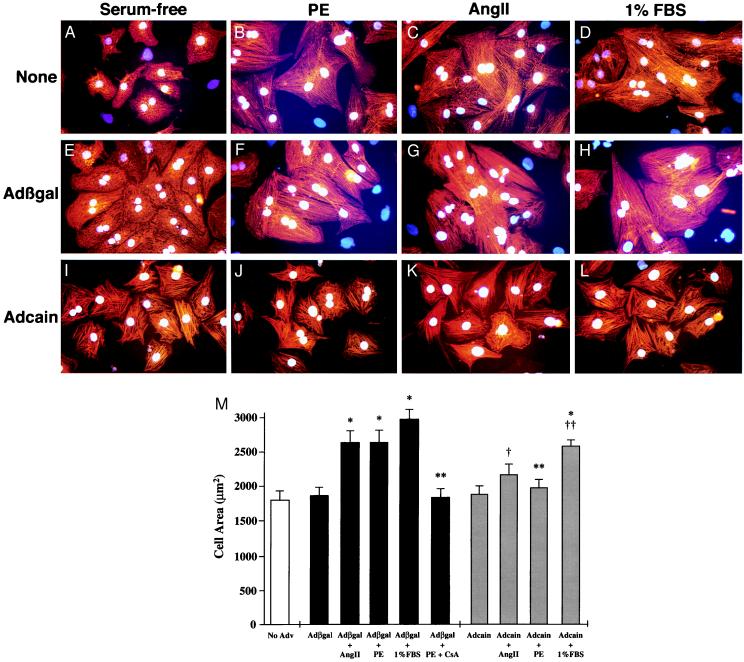
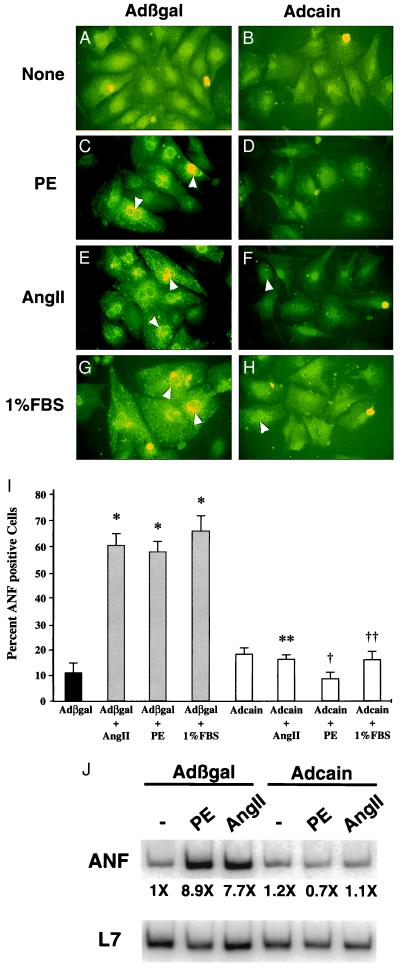
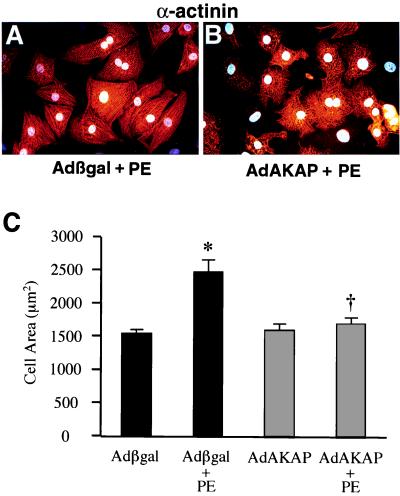
Cardiac hypertrophy is a major predictor of future morbidity and mortality. Recent investigation has centered around identifying the molecular signaling pathways that regulate cardiac myocyte reactivity with the goal of modulating pathologic hypertrophic programs. One potential regulator of cardiomyocyte hypertrophy is the calcium-sensitive phosphatase calcineurin. We show here that calcineurin enzymatic activity, mRNA, and protein levels are increased in cultured neonatal rat cardiomyocytes by hypertrophic agonists such as angiotensin II, phenylephrine, and 1% fetal bovine serum. This induction of calcineurin activity was associated with an increase in calcineurin Abeta (CnAbeta) mRNA and protein, but not in CnAalpha or CnAgamma. Agonist-dependent increases in calcineurin enzymatic activity were specifically inhibited with an adenovirus expressing a noncompetitive peptide inhibitor of calcineurin known as cain [Lai, M. M., Burnett, P. E., Wolosker, H., Blackshaw, S. & Snyder, S. H. (1998) J. Biol. Chem. 273, 18325-18331]. Targeted inhibition of calcineurin with cain or an adenovirus expressing only the calcineurin inhibitory domain of AKAP79 attenuated cardiomyocyte hypertrophy and atrial natriuretic factor expression in response to angiotensin II, phenylephrine, and 1% fetal bovine serum. These data demonstrate that calcineurin is an important regulator of cardiomyocyte hypertrophy in response to certain agonists and suggest that cyclosporin A and FK506 function to attenuate cardiac hypertrophy by specifically inhibiting calcineurin.
Figures
References
-
- Levy D, Garrison R J, Savage D D, Kannel W B, Castelli W P. N Engl J Med. 1990;322:1561–1566. - PubMed
-
- Sadoshima J, Izumo S. Annu Rev Physiol. 1997;59:551–571. - PubMed
-
- Dorn G W, II, Brown J H. Trends Cardiovasc Med. 1999;9:26–34. - PubMed
-
- Sugden P H. Circ Res. 1999;84:633–646. - PubMed
-
- Olson E N, Molkentin J D. Circ Res. 1999;84:623–632. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
