

Caspase-2 mediates neuronal cell death induced by beta-amyloid
- PMID: 10662829
- PMCID: PMC6772358
- DOI: 10.1523/JNEUROSCI.20-04-01386.2000
Caspase-2 mediates neuronal cell death induced by beta-amyloid
Abstract
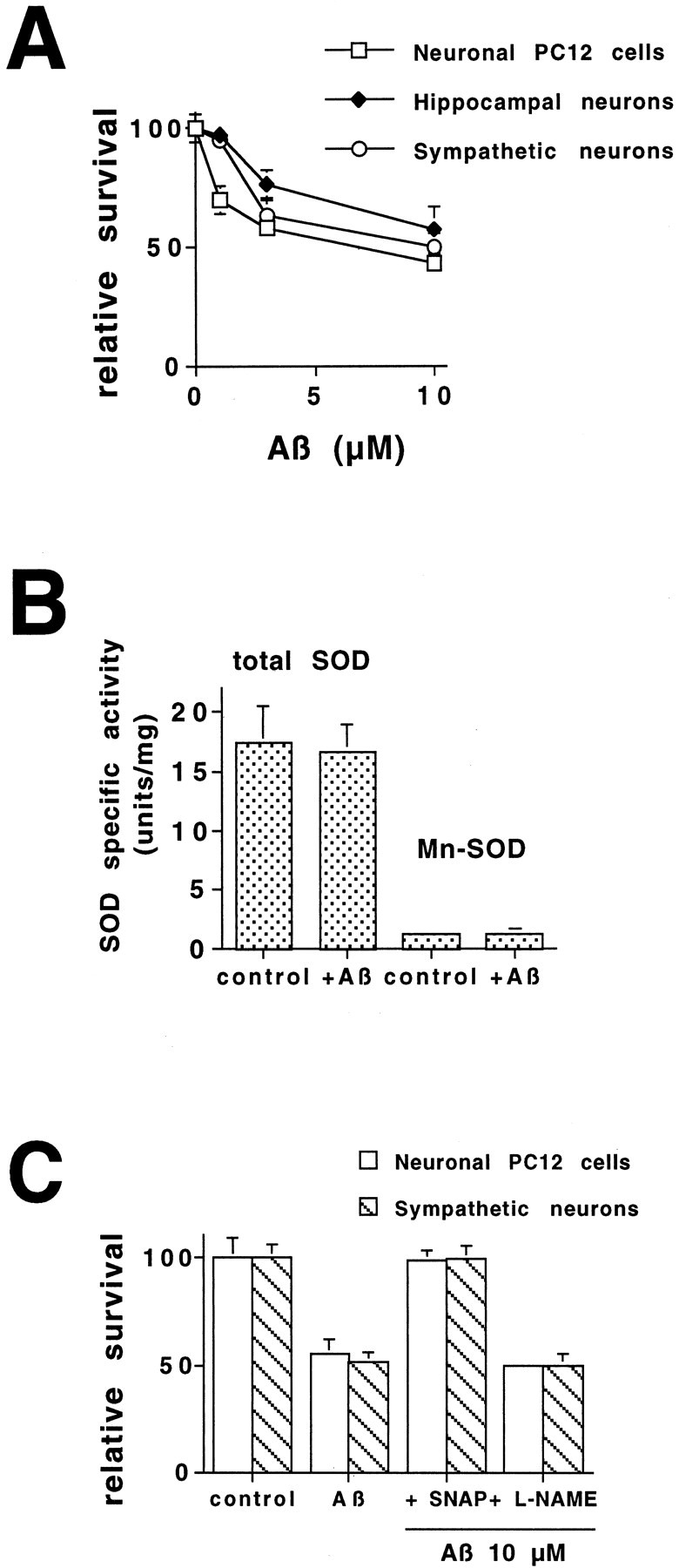
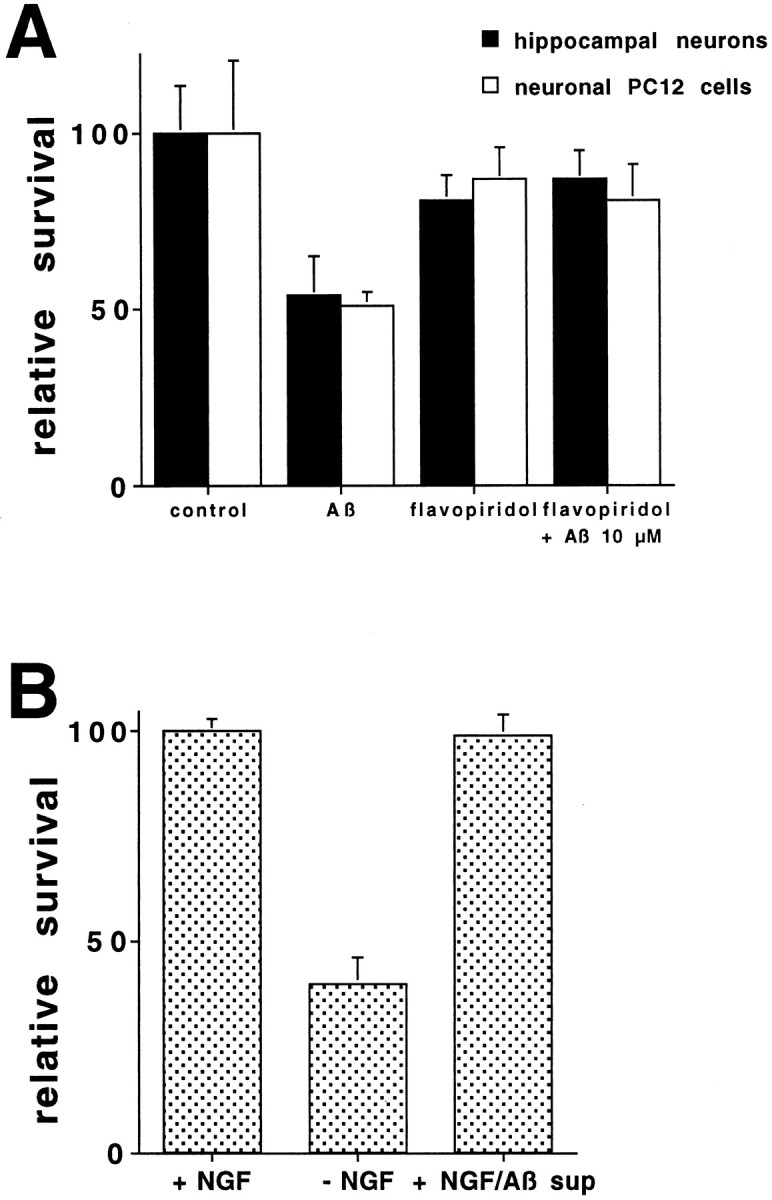
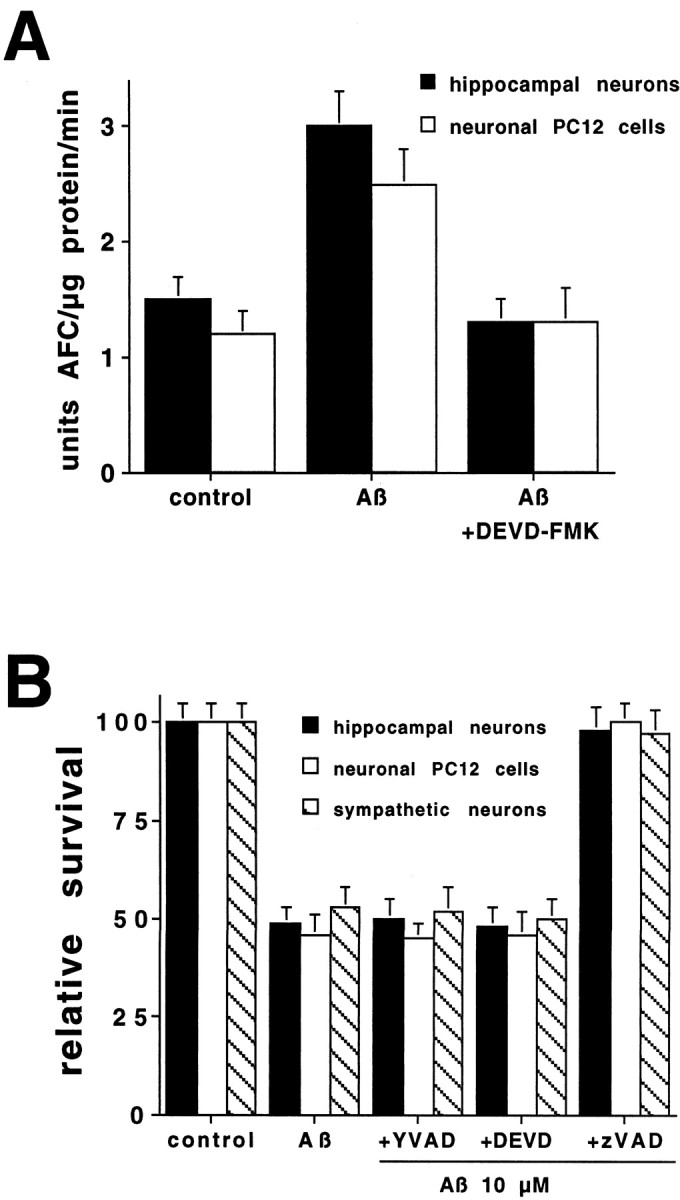
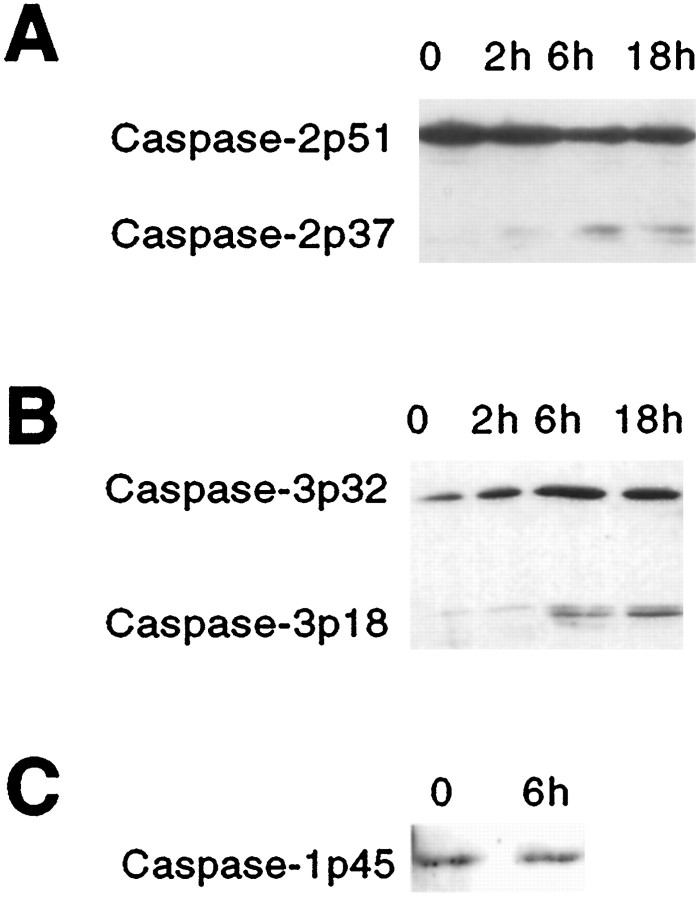
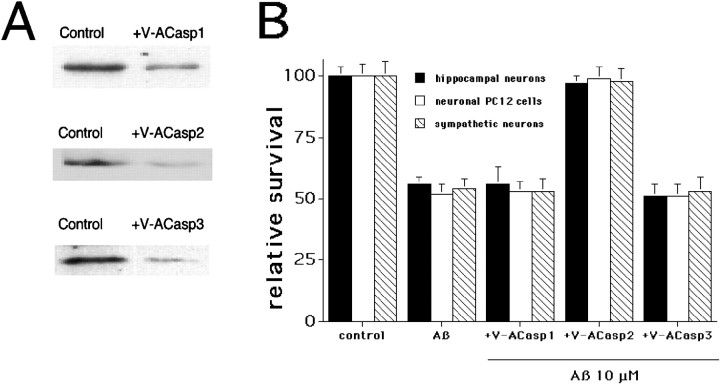
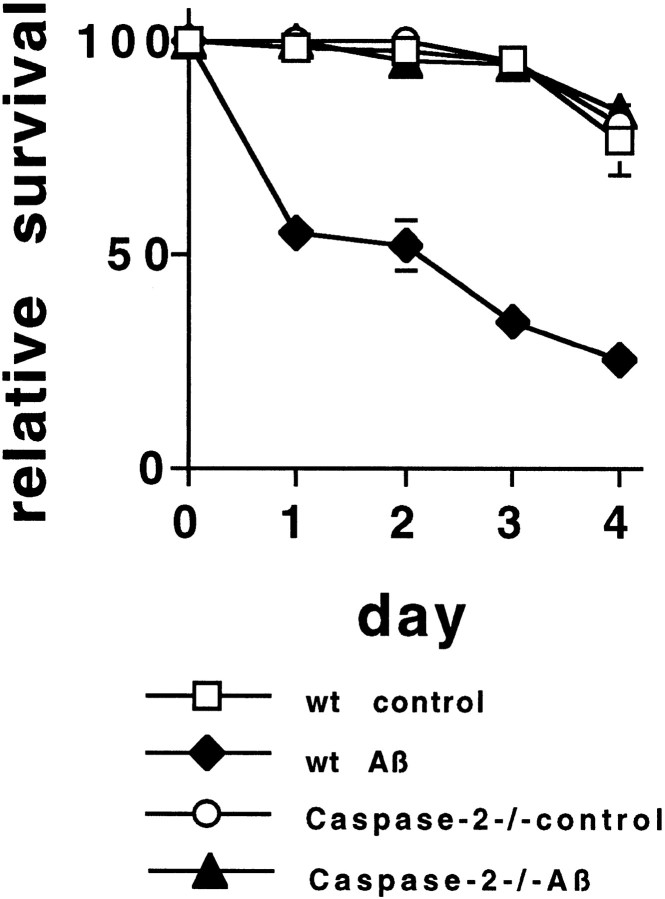
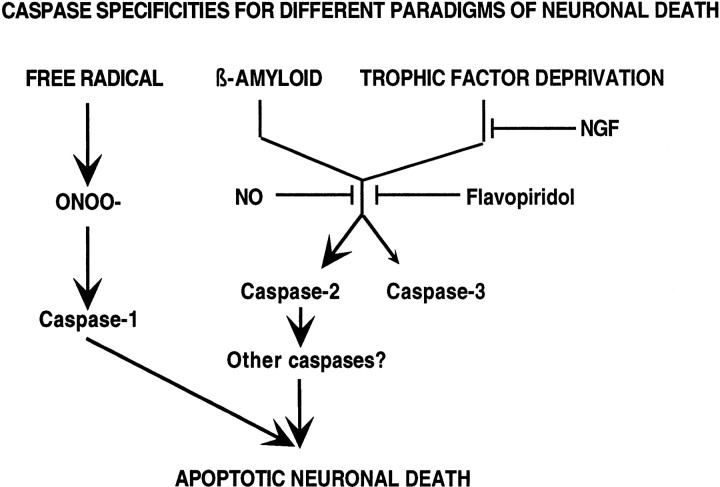
beta-amyloid (Abeta) has been proposed to play a role in the pathogenesis of Alzheimer's disease (AD). Deposits of insoluble Abeta are found in the brains of patients with AD and are one of the pathological hallmarks of the disease. It has been proposed that Abeta induces death by oxidative stress, possibly through the generation of peroxynitrite from superoxide and nitric oxide. In our current study, treatment with nitric oxide generators protected against Abeta-induced death, whereas inhibition of nitric oxide synthase afforded no protection, suggesting that formation of peroxynitrite is not critical for Abeta-mediated death. Previous studies have shown that aggregated Abeta can induce caspase-dependent apoptosis in cultured neurons. In all of the neuronal populations studied here (hippocampal neurons, sympathetic neurons, and PC12 cells), cell death was blocked by the broad spectrum caspase inhibitor N-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone and more specifically by the downregulation of caspase-2 with antisense oligonucleotides. In contrast, downregulation of caspase-1 or caspase-3 did not block Abeta(1-42)-induced death. Neurons from caspase-2 null mice were totally resistant to Abeta(1-42) toxicity, confirming the importance of this caspase in Abeta-induced death. The results indicate that caspase-2 is necessary for Abeta(1-42)-induced apoptosis in vitro.
Figures
References
-
- Ahmad M, Srinivasula SM, Hegde R, Mukattash R, Fernandes-Alnemri T, Alnemri ES. Identification and characterization of murine caspase-14, a new member of the caspase family. Cancer Res. 1998;58:5201–5205. - PubMed
-
- Chan SL, Griffin WS, Mattson MP. Evidence for caspase-mediated cleavage of AMPA receptor subunits in neuronal apoptosis and Alzheimer's disease. J Neurosci Res. 1999;57:315–323. - PubMed
-
- Cotman CW, Su JH. Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol. 1996;6:493–506. - PubMed
-
- Cotman CW, Whittemore ER, Watt JA, Anderson AJ, Loo DT. Possible role of apoptosis in Alzheimer's disease. Ann NY Acad Sci. 1994;747:36–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials