

Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway
- PMID: 10669763
- PMCID: PMC85374
- DOI: 10.1128/MCB.20.5.1886-1896.2000
Posttranslational modification of Bcl-2 facilitates its proteasome-dependent degradation: molecular characterization of the involved signaling pathway
Abstract
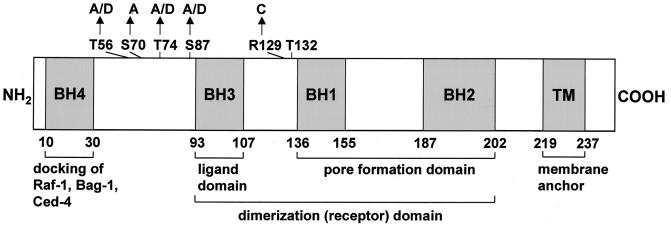
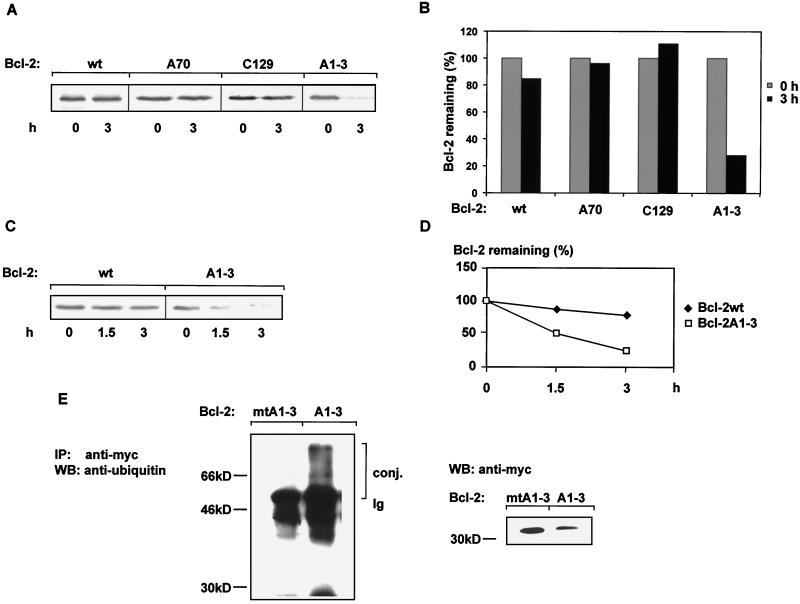
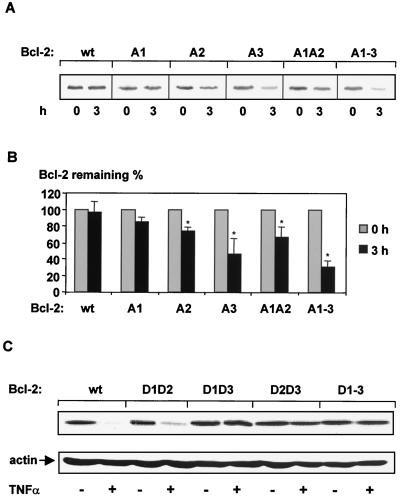
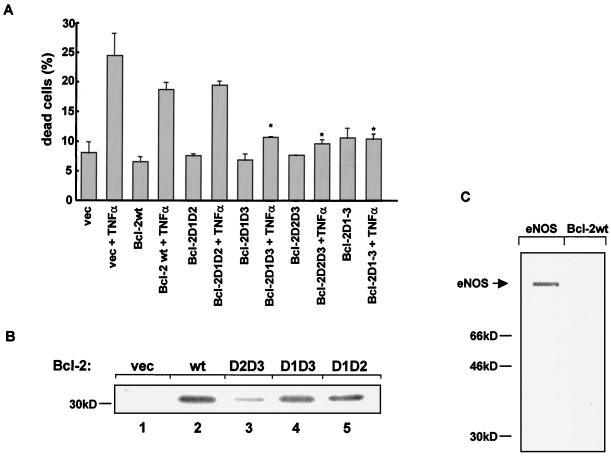
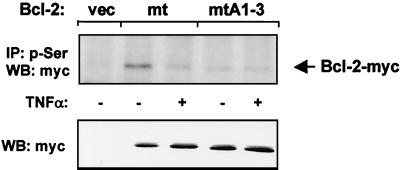
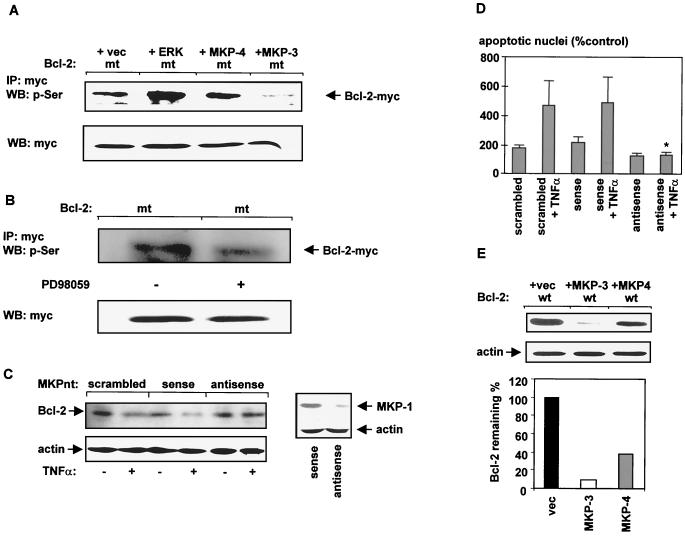
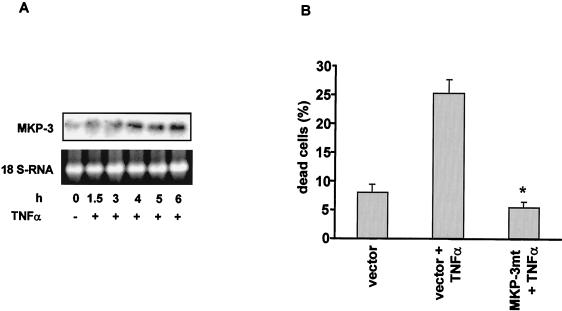
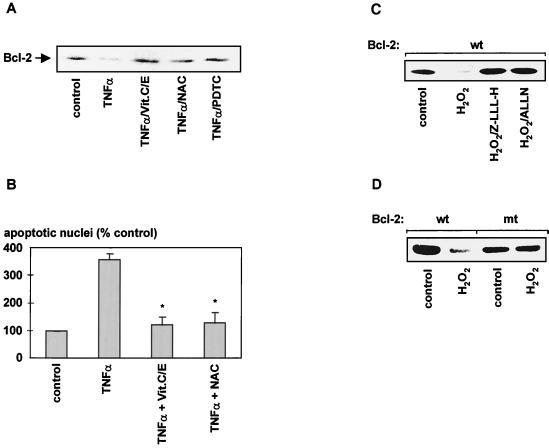
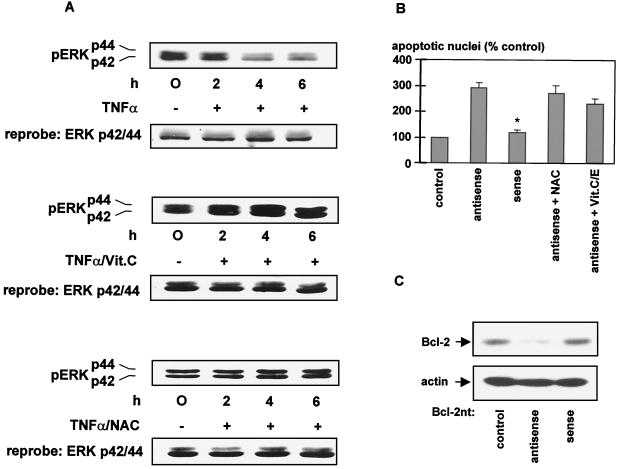
The ratio of proapoptotic versus antiapoptotic Bcl-2 members is a critical determinant that plays a significant role in altering susceptibility to apoptosis. Therefore, a reduction of antiapoptotic protein levels in response to proximal signal transduction events may switch on the apoptotic pathway. In endothelial cells, tumor necrosis factor alpha (TNF-alpha) induces dephosphorylation and subsequent ubiquitin-dependent degradation of the antiapoptotic protein Bcl-2. Here, we investigate the role of different putative phosphorylation sites to facilitate Bcl-2 degradation. Mutation of the consensus protein kinase B/Akt site or of potential protein kinase C or cyclic AMP-dependent protein kinase sites does not affect Bcl-2 stability. In contrast, inactivation of the three consensus mitogen-activated protein (MAP) kinase sites leads to a Bcl-2 protein that is ubiquitinated and subsequently degraded by the 26S proteasome. Inactivation of these sites within Bcl-2 revealed that dephosphorylation of Ser87 appears to play a major role. A Ser-to-Ala substitution at this position results in 50% degradation, whereas replacement of Thr74 with Ala leads to 25% degradation, as assessed by pulse-chase studies. We further demonstrated that incubation with TNF-alpha induces dephosphorylation of Ser87 of Bcl-2 in intact cells. Furthermore, MAP kinase triggers phosphorylation of Bcl-2, whereas a reduction in Bcl-2 phosphorylation was observed in the presence of MAP kinase-specific phosphatases or the MAP kinase-specific inhibitor PD98059. Moreover, we show that oxidative stress mediates TNF-alpha-stimulated proteolytic degradation of Bcl-2 by reducing MAP kinase activity. Taken together, these results demonstrate a direct protective role for Bcl-2 phosphorylation by MAP kinase against apoptotic challenges to endothelial cells and other cells.
Figures
References
-
- Adams J M, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1325. - PubMed
-
- Arch R H, Gedrich R W, Thompson C B. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Dev. 1998;12:2821–2830. - PubMed
-
- Ashkenazi A, Dixit V M. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. - PubMed
-
- Baffy G, Miyashita T, Williamson J R, Reed J C. Apoptosis induced by withdrawal of interleukin-3 (IL-3) from an IL-3-dependent hemapoietic cell line is associated with repartitioning of intracellular calcium and is blocked by enforced Bcl-2 oncoprotein production. J Biol Chem. 1993;268:6511–6519. - PubMed
-
- Batt D B, Carmichael G G, Liu Z. An improved rapid method of isolating RNA from cultured cells. Methods Mol Biol. 1998;86:15–17. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases