

Methionine adenosyltransferase I/III deficiency: novel mutations and clinical variations
- PMID: 10677294
- PMCID: PMC1288087
- DOI: 10.1086/302752
Methionine adenosyltransferase I/III deficiency: novel mutations and clinical variations
Abstract
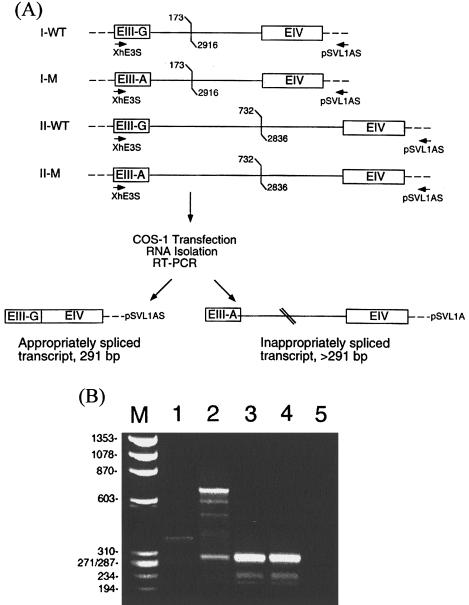
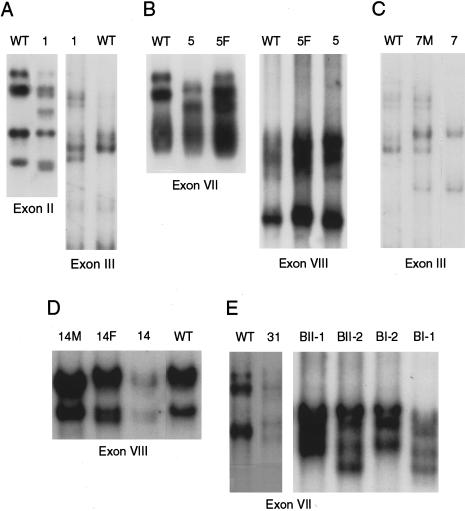
Methionine adenosyltransferase (MAT) I/III deficiency, caused by mutations in the MAT1A gene, is characterized by persistent hypermethioninemia without elevated homocysteine or tyrosine. Clinical manifestations are variable and poorly understood, although a number of individuals with homozygous null mutations in MAT1A have neurological problems, including brain demyelination. We analyzed MAT1A in seven hypermethioninemic individuals, to provide insight into the relationship between genotype and phenotype. We identified six novel mutations and demonstrated that mutations resulting in high plasma methionines may signal clinical difficulties. Two patients-a compound heterozygote for truncating and severely inactivating missense mutations and a homozygote for an aberrant splicing MAT1A mutation-have plasma methionine in the 1,226-1,870 microM range (normal 5-35 microM) and manifest abnormalities of the brain gray matter or signs of brain demyelination. Another compound heterozygote for truncating and inactivating missense mutations has 770-1,240 microM plasma methionine and mild cognitive impairment. Four individuals carrying either two inactivating missense mutations or the single-allelic R264H mutation have 105-467 microM plasma methionine and are clinically unaffected. Our data underscore the necessity of further studies to firmly establish the relationship between genotypes in MAT I/III deficiency and clinical phenotypes, to elucidate the molecular bases of variability in manifestations of MAT1A mutations.
Figures

References
-
- Blom HJ, Davidson AJ, Finkelstein JD, Luder AS, Bernardini I, Martin JJ, Tangerman A, et al (1992) Persistent hypermethioninaemia with dominant inheritance. J Inherit Metab Dis 15:188–197 - PubMed
-
- Cantoni GL (1953) S-adenosylmethionine: a new intermediate formed enzymatically from L-methionine and adenosinetriphosphate. J Biol Chem 204:403–416 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
