

Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene
- PMID: 10677295
- PMCID: PMC1288088
- DOI: 10.1086/302751
Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene
Abstract
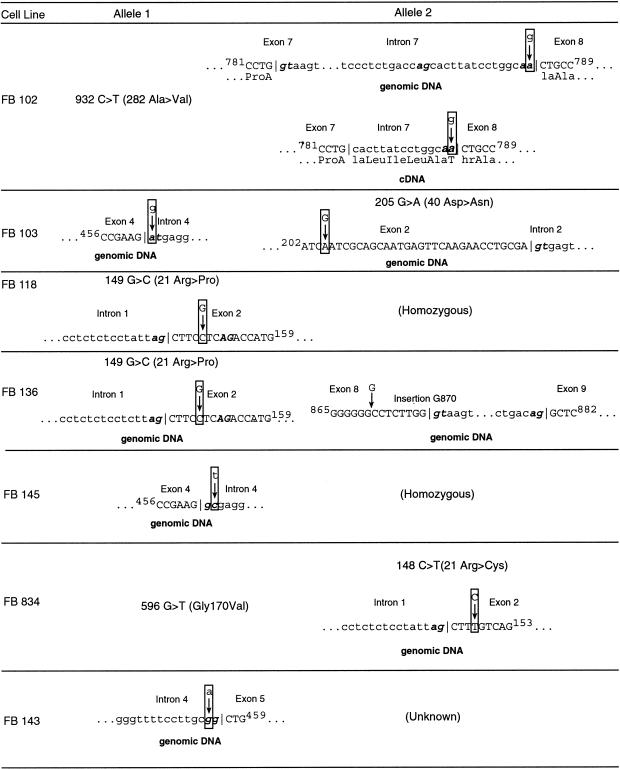
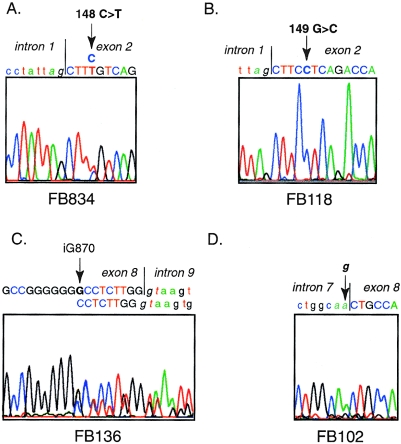
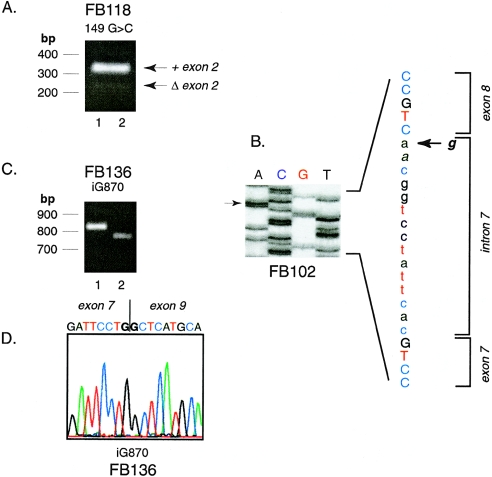
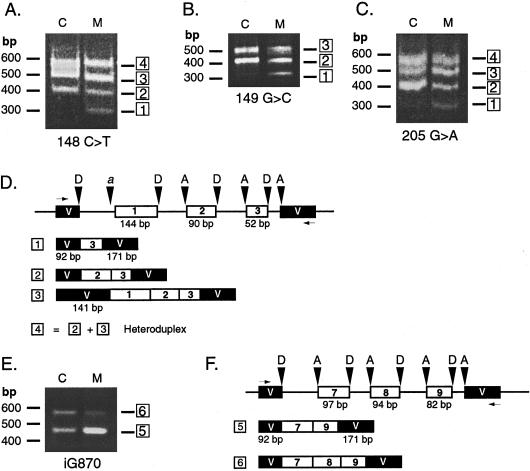
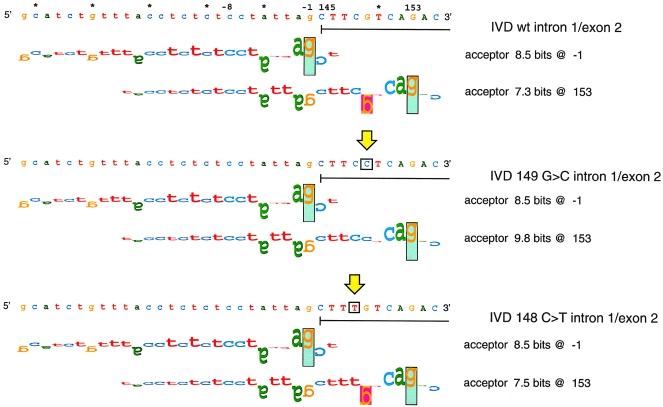
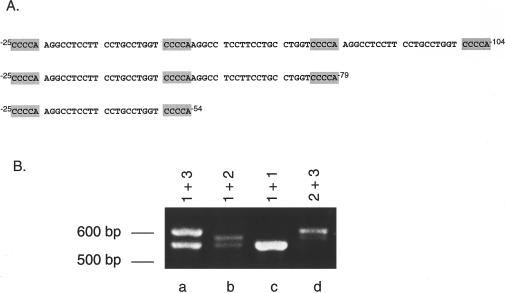
Isovaleric acidemia (IVA) is a recessive disorder caused by a deficiency of isovaleryl-CoA dehydrogenase (IVD). We have reported elsewhere nine point mutations in the IVD gene in fibroblasts of patients with IVA, which lead to abnormalities in IVD protein processing and activity. In this report, we describe eight IVD gene mutations identified in seven IVA patients that result in abnormal splicing of IVD RNA. Four mutations in the coding region lead to aberrantly spliced mRNA species in patient fibroblasts. Three of these are amino acid altering point mutations, whereas one is a single-base insertion that leads to a shift in the reading frame of the mRNA. Two of the coding mutations strengthen pre-existing cryptic splice acceptors adjacent to the natural splice junctions and apparently interfere with exon recognition, resulting in exon skipping. This mechanism for missplicing has not been reported elsewhere. Four other mutations alter either the conserved gt or ag dinucleotide splice sites in the IVD gene. Exon skipping and cryptic splicing were confirmed by transfection of these mutations into a Cos-7 cell line model splicing system. Several of the mutations were predicted by individual information analysis to inactivate or significantly weaken adjacent donor or acceptor sites. The high frequency of splicing mutations identified in these patients is unusual, as is the finding of missplicing associated with missense mutations in exons. These results may lead to a better understanding of the phenotypic complexity of IVA, as well as provide insight into those factors important in defining intron/exon boundaries in vivo.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank (accession numbers AF191214 to AF191218)
-
- Laboratory of Computational and Experimental Biology at the National Institutes of Health, http://www.lecb.ncifcrf.gov/~toms/ (for the Scan program)
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for IVA [MIM 243500])
References
-
- Berg MA, Guevara-Aguirre J, Rosenbloom AL, Rosenfeld RG, Francke U (1992) Mutation creating a new splice site in the growth hormone receptor genes of 37 Ecuadorean patients with Laron syndrome. Hum Mutat 1:24–32 - PubMed
-
- Crane F, Beinert H (1955) On the mechanism of dehydrogenation of fatty acyl derivatives of coenzyme A: part II. The electron-transferring flavoproteins. J Biol Chem 218:717–731 - PubMed
-
- Dietz HC, Kendzior RJ Jr. (1994) Maintenance of an open reading frame as an additional level of scrutiny during splice site selection. Nat Genet 8:183–188 - PubMed
-
- Dietz HC, Valle D, Francomano CA, Kendzior RJ Jr., Pyeritz RE, Cutting GR (1993) The skipping of constitutive exons in vivo induced by nonsense mutations. Science 259:680–683 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
