

Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene
- PMID: 10677296
- PMCID: PMC1288089
- DOI: 10.1086/302750
Autosomal recessive disorder otospondylomegaepiphyseal dysplasia is associated with loss-of-function mutations in the COL11A2 gene
Abstract
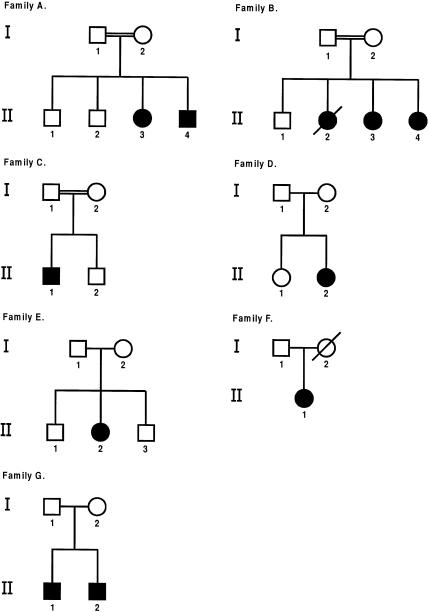
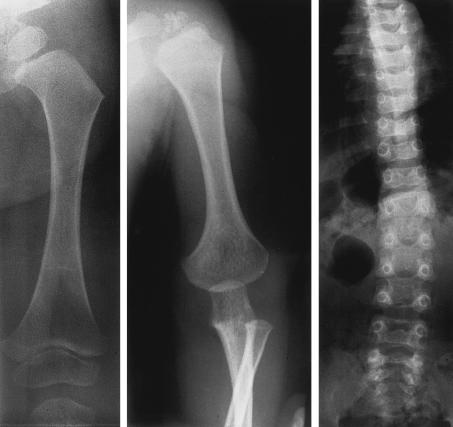
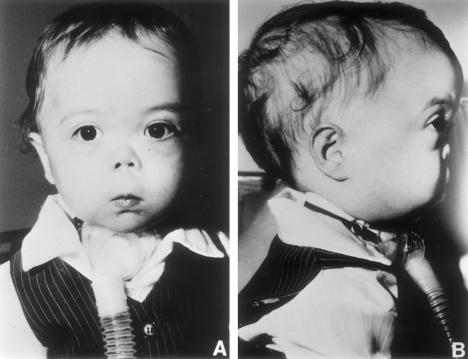
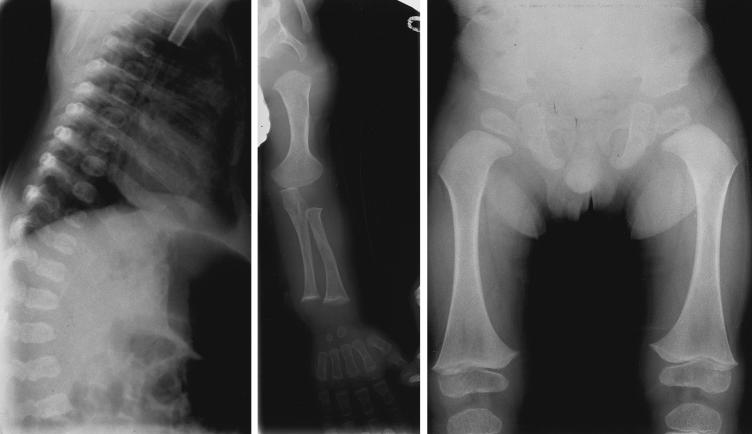
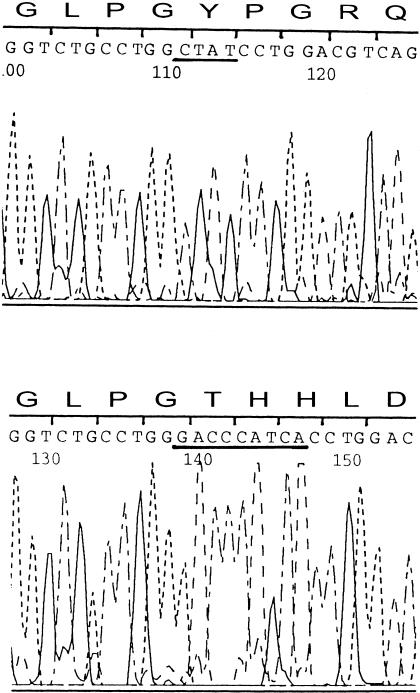
Otospondylomegaepiphyseal dysplasia (OSMED) is an autosomal recessive skeletal dysplasia accompanied by severe hearing loss. The phenotype overlaps that of the autosomal dominant disorders-Stickler and Marshall syndromes-but can be distinguished by disproportionately short limbs, severe hearing loss, and lack of ocular involvement. In one family with OSMED, a homozygous Gly-->Arg substitution has been described in COL11A2, which codes for the alpha2 chain of type XI collagen. We report seven further families with OSMED. All affected individuals had a remarkably similar phenotype: profound sensorineural hearing loss, skeletal dysplasia with limb shortening and large epiphyses, cleft palate, an extremely flat face, hypoplasia of the mandible, a short nose with anteverted nares, and a flat nasal bridge. We screened affected individuals for mutations in COL11A2 and found different mutations in each family. Individuals from four families, including three with consanguineous parents, were homozygous for mutations. Individuals from three other families, in whom parents were nonconsanguineous, were compound heterozygous. Of the 10 identified mutations, 9 are predicted to cause premature termination of translation, and 1 is predicted to cause an in-frame deletion. We conclude that the OSMED phenotype is highly homogenous and results from homozygosity or compound heterozygosity for COL11A2 mutations, most of which are predicted to cause complete absence of alpha2(XI) chains.
Figures
Similar articles
-
COL11A2 mutation associated with autosomal recessive Weissenbacher-Zweymuller syndrome: molecular and clinical overlap with otospondylomegaepiphyseal dysplasia (OSMED).Am J Med Genet A. 2005 Jan 1;132A(1):33-5. doi: 10.1002/ajmg.a.30371. Am J Med Genet A. 2005. PMID: 15558753
-
Oto-spondylo-megaepiphyseal dysplasia (OSMED): clinical and radiological findings in sibs homozygous for premature stop codon mutation in the COL11A2 gene.Am J Med Genet A. 2006 Jun 1;140(11):1189-95. doi: 10.1002/ajmg.a.31205. Am J Med Genet A. 2006. PMID: 16637051
-
Oto- spondylo-megaepiphyseal dysplasia (OSMED): clinical description of three patients homozygous for a missense mutation in the COL11A2 gene.Am J Med Genet. 1997 Jun 13;70(3):315-23. doi: 10.1002/(sici)1096-8628(19970613)70:3<315::aid-ajmg19>3.3.co;2-y. Am J Med Genet. 1997. PMID: 9188673
-
The type XI collagenopathies.Pediatr Radiol. 1998 Oct;28(10):745-50. doi: 10.1007/s002470050459. Pediatr Radiol. 1998. PMID: 9799295 Review.
-
A stop codon mutation in COL11A2 induces exon skipping and leads to non-ocular Stickler syndrome.Am J Med Genet A. 2004 Oct 1;130A(2):160-4. doi: 10.1002/ajmg.a.30111. Am J Med Genet A. 2004. PMID: 15372529 Review.
Cited by
-
A type II collagen mutation also results in oto-spondylo-megaepiphyseal dysplasia.Hum Genet. 2005 Nov;118(2):175-8. doi: 10.1007/s00439-005-0058-0. Epub 2005 Nov 15. Hum Genet. 2005. PMID: 16189708
-
Gene-disease network analysis reveals functional modules in mendelian, complex and environmental diseases.PLoS One. 2011;6(6):e20284. doi: 10.1371/journal.pone.0020284. Epub 2011 Jun 14. PLoS One. 2011. PMID: 21695124 Free PMC article.
-
Dominant and recessive forms of fibrochondrogenesis resulting from mutations at a second locus, COL11A2.Am J Med Genet A. 2012 Feb;158A(2):309-14. doi: 10.1002/ajmg.a.34406. Epub 2012 Jan 13. Am J Med Genet A. 2012. PMID: 22246659 Free PMC article.
-
Molecular Basis of Pathogenic Variants in the Fibrillar Collagens.Genes (Basel). 2022 Jul 4;13(7):1199. doi: 10.3390/genes13071199. Genes (Basel). 2022. PMID: 35885981 Free PMC article. Review.
-
Minor fibrillar collagens, variable regions alternative splicing, intrinsic disorder, and tyrosine sulfation.Protein Cell. 2012 Jun;3(6):419-33. doi: 10.1007/s13238-012-2917-5. Epub 2012 Jul 1. Protein Cell. 2012. PMID: 22752873 Free PMC article. Review.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.nbci.nlm.nih.gov/Omim (for OSMED [215150], Stickler syndrome types I [108300] and II [184840], Marshall syndrome [154780], and Weissenbacher-Zweymüller syndrome [277610])
References
-
- Ala-Kokko L, Kvist A-P, Metsäranta M, Kivirikko KI, de Crombrugghe B, Prockop DJ, Vuorio E (1995) Conservation of the sizes of 53 introns and over 100 intronic sequences for the binding of common transcription factors in the human and mouse genes for type II procollagen (COL2A1). Biochem J 308:923–929 - PMC - PubMed
-
- Brewton RG, Mayne R (1994) Heterotypic type II, IX and XI fibrils: comparison of vitreous and cartilage forms. In: Yurchenco PD, Birk DE, Mecham RP (eds) Extracellular matrix assembly and structure. Academic Press, San Diego, pp 129–170
-
- Cremer MA, Rosloniec EF, Kang AH (1998) The cartilage collagens: a review of their structure, organization, and role in the pathogenesis of experimental arthritis in animals and in human rheumatic disease. J Mol Med 76:275–288 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
