

Familial syndromic esophageal atresia maps to 2p23-p24
- PMID: 10677303
- PMCID: PMC1288096
- DOI: 10.1086/302779
Familial syndromic esophageal atresia maps to 2p23-p24
Abstract
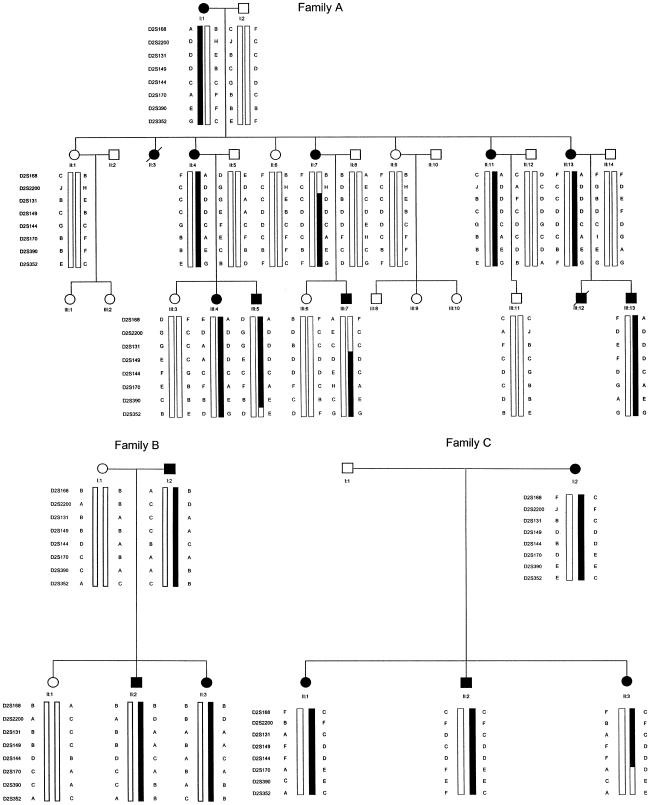
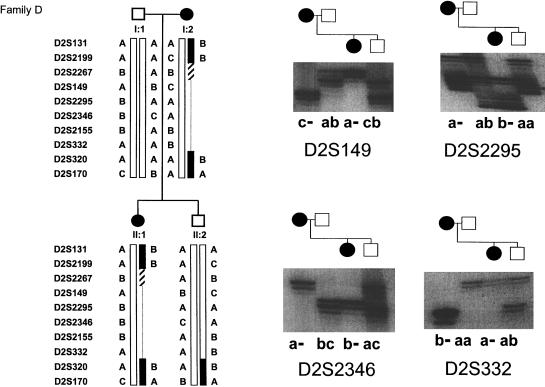
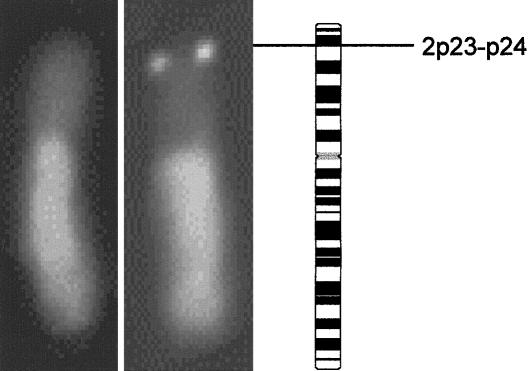
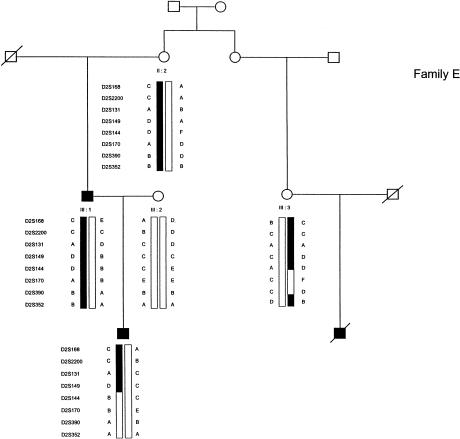
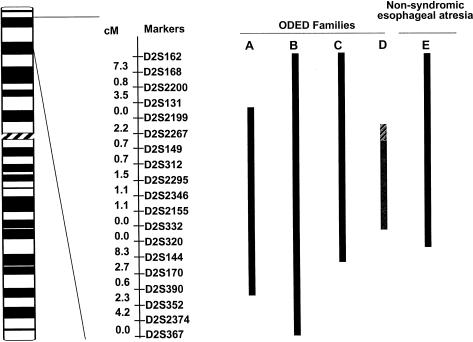
Esophageal atresia (EA) is a common life-threatening congenital anomaly that occurs in 1/3,000 newborns. Little is known of the genetic factors that underlie EA. Oculodigitoesophageoduodenal (ODED) syndrome (also known as "Feingold syndrome") is a rare autosomal dominant disorder with digital abnormalities, microcephaly, short palpebral fissures, mild learning disability, and esophageal/duodenal atresia. We studied four pedigrees, including a three-generation Dutch family with 11 affected members. Linkage analysis was initially aimed at chromosomal regions harboring candidate genes for this disorder. Twelve different genomic regions covering 15 candidate genes (approximately 15% of the genome) were excluded from involvement in the ODED syndrome. A subsequent nondirective mapping approach revealed evidence for linkage between the syndrome and marker D2S390 (maximum LOD score 4.51 at recombination fraction 0). A submicroscopic deletion in a fourth family with ODED provided independent confirmation of this genetic localization and narrowed the critical region to 7.3 cM in the 2p23-p24 region. These results show that haploinsufficiency for a gene or genes in 2p23-p24 is associated with syndromic EA.
Figures

References
Electronic-Database Information
-
- Généthon, http://www.genethon.fr/
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for VATER association [MIM 192350] and ODED syndrome [MIM 164280])
-
- Stanford Human Genome Center, http://www-shgc.stanford.edu/
-
- Whitehead Institute for Biomedical Research/MIT Center for Genome Research, http://www-genome.wi.mit.edu/
References
-
- Båvik C, Ward SJ, Ong DE (1997) Identification of a mechanism to localize generation of retinoic acid in rat embryos. Mech Dev 69:155–167 - PubMed
-
- Beasley SW, Allen M, Myers N (1997) The effects of Down syndrome and other chromosomal abnormalities on survival and management in esophageal atresia. Pediatr Surg Int 12:550–551 - PubMed
-
- Boles RG, Pober BR, Gibson LH, Willis CR, McGrath J, Roberts DJ, Yang Feng TL (1995) Deletion of chromosome 2q24-q31 causes characteristic digital anomalies: case report and review. Am J Med Genet 55:155–160 - PubMed
-
- Boulet AM, Capecchi MR (1996) Targeted disruption of Hoxc 4 causes esophageal defects and vertebral transformations. Dev Biol 177:232–249 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
