

Comparative genome analysis of the pathogenic spirochetes Borrelia burgdorferi and Treponema pallidum
- PMID: 10678983
- PMCID: PMC97324
- DOI: 10.1128/IAI.68.3.1633-1648.2000
Comparative genome analysis of the pathogenic spirochetes Borrelia burgdorferi and Treponema pallidum
Abstract
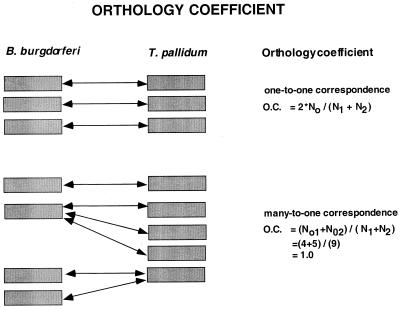
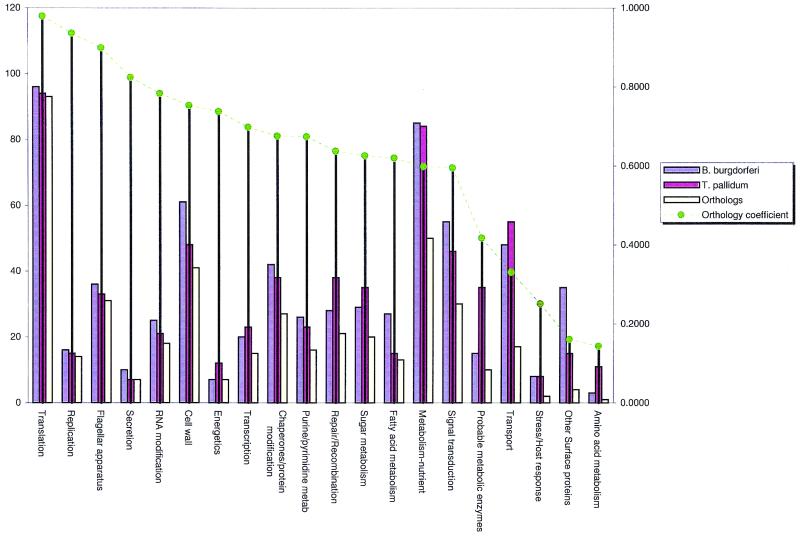
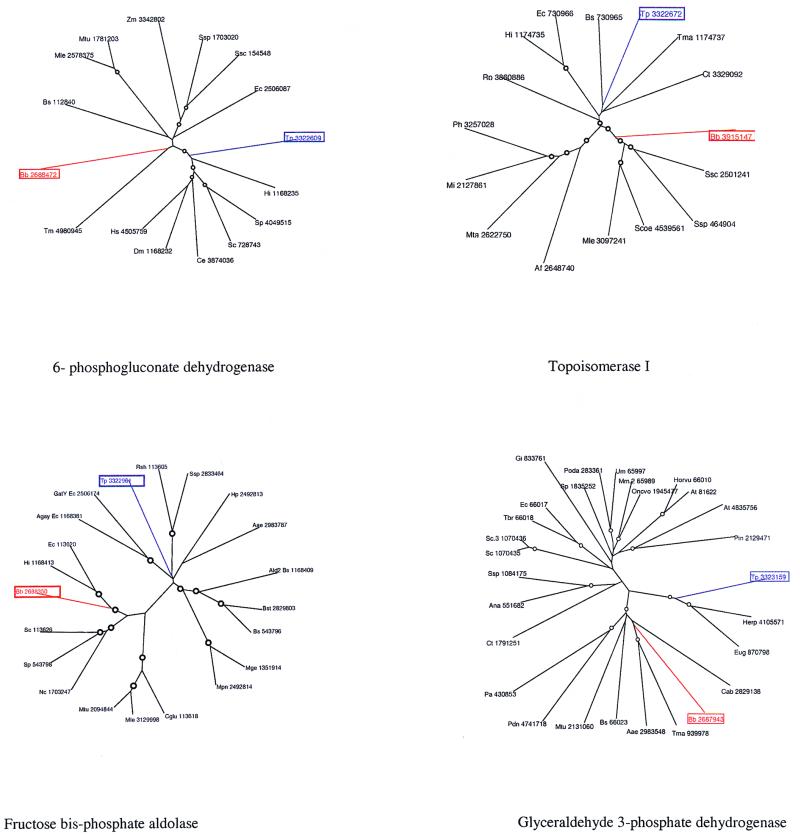
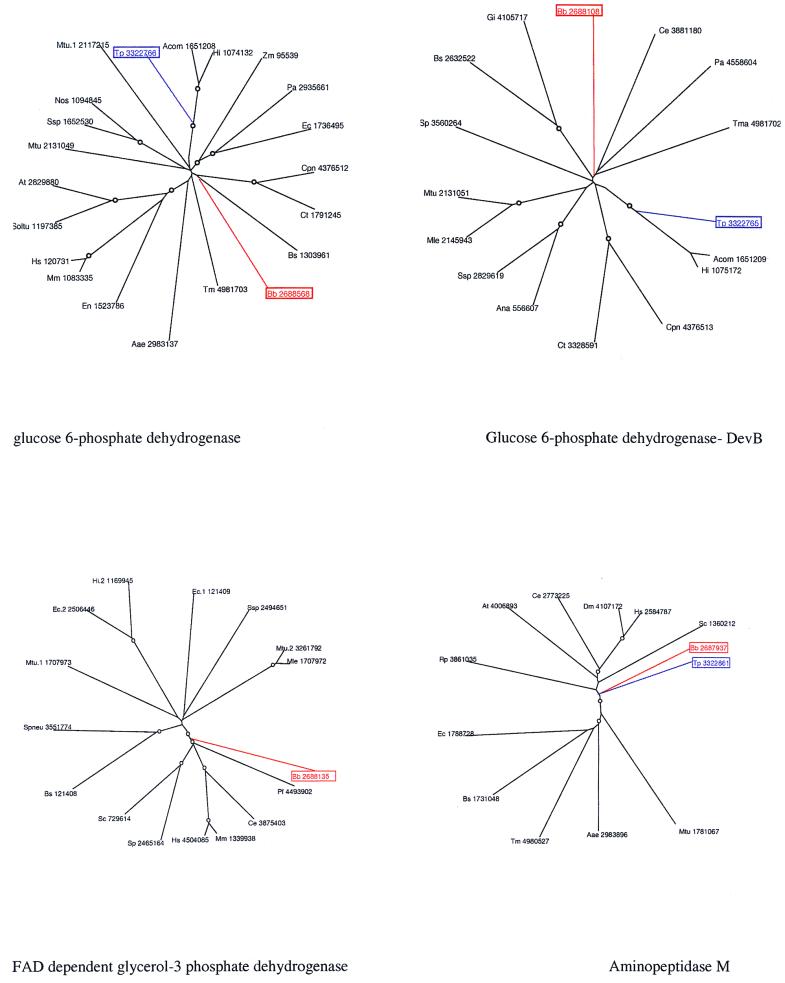
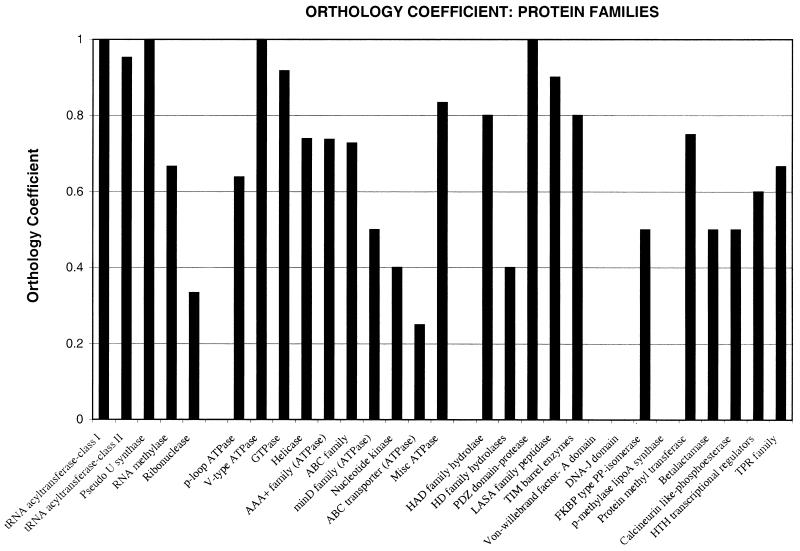
A comparative analysis of the predicted protein sequences encoded in the complete genomes of Borrelia burgdorferi and Treponema pallidum provides a number of insights into evolutionary trends and adaptive strategies of the two spirochetes. A measure of orthologous relationships between gene sets, termed the orthology coefficient (OC), was developed. The overall OC value for the gene sets of the two spirochetes is about 0.43, which means that less than one-half of the genes show readily detectable orthologous relationships. This emphasizes significant divergence between the two spirochetes, apparently driven by different biological niches. Different functional categories of proteins as well as different protein families show a broad distribution of OC values, from near 1 (a perfect, one-to-one correspondence) to near 0. The proteins involved in core biological functions, such as genome replication and expression, typically show high OC values. In contrast, marked variability is seen among proteins that are involved in specific processes, such as nutrient transport, metabolism, gene-specific transcription regulation, signal transduction, and host response. Differences in the gene complements encoded in the two spirochete genomes suggest active adaptive evolution for their distinct niches. Comparative analysis of the spirochete genomes produced evidence of gene exchanges with other bacteria, archaea, and eukaryotic hosts that seem to have occurred at different points in the evolution of the spirochetes. Examples are presented of the use of sequence profile analysis to predict proteins that are likely to play a role in pathogenesis, including secreted proteins that contain specific protein-protein interaction domains, such as von Willebrand A, YWTD, TPR, and PR1, some of which hitherto have been reported only in eukaryotes. We tentatively reconstruct the likely evolutionary process that has led to the divergence of the two spirochete lineages; this reconstruction seems to point to an ancestral state resembling the symbiotic spirochetes found in insect guts.
Figures
Similar articles
-
Genome analyses of spirochetes: a study of the protein structures, functions and metabolic pathways in Treponema pallidum and Borrelia burgdorferi.J Mol Microbiol Biotechnol. 2000 Oct;2(4):387-92. J Mol Microbiol Biotechnol. 2000. PMID: 11075910
-
Complete genome sequence of Treponema pallidum, the syphilis spirochete.Science. 1998 Jul 17;281(5375):375-88. doi: 10.1126/science.281.5375.375. Science. 1998. PMID: 9665876
-
Whole genome analyses of transporters in spirochetes: Borrelia burgdorferi and Treponema pallidum.J Mol Microbiol Biotechnol. 2000 Oct;2(4):393-9. J Mol Microbiol Biotechnol. 2000. PMID: 11075911
-
Interaction of spirochetes with the host.Res Microbiol. 1992 Jul-Aug;143(6):629-39. doi: 10.1016/0923-2508(92)90121-4. Res Microbiol. 1992. PMID: 1475523 Review.
-
Genetic approaches to cell biology and metabolism of spirochetes.Res Microbiol. 1992 Jul-Aug;143(6):605-13. doi: 10.1016/0923-2508(92)90118-8. Res Microbiol. 1992. PMID: 1475521 Review.
Cited by
-
Genomic insights into the c-di-GMP signaling and biofilm development in the saprophytic spirochete Leptospira biflexa.Arch Microbiol. 2023 Apr 8;205(5):180. doi: 10.1007/s00203-023-03519-7. Arch Microbiol. 2023. PMID: 37031284
-
Genomic survey and expression analysis of DNA repair genes in the genus Leptospira.Mol Genet Genomics. 2016 Apr;291(2):703-22. doi: 10.1007/s00438-015-1135-2. Epub 2015 Nov 2. Mol Genet Genomics. 2016. PMID: 26527082
-
Investigation of the immune escape mechanism of Treponema pallidum.Infection. 2023 Apr;51(2):305-321. doi: 10.1007/s15010-022-01939-z. Epub 2022 Oct 19. Infection. 2023. PMID: 36260281 Review.
-
Weighted genome trees: refinements and applications.J Bacteriol. 2005 Feb;187(4):1305-16. doi: 10.1128/JB.187.4.1305-1316.2005. J Bacteriol. 2005. PMID: 15687194 Free PMC article.
-
Phosphoenolpyruvate Phosphotransferase System Components Modulate Gene Transcription and Virulence of Borrelia burgdorferi.Infect Immun. 2015 Dec 28;84(3):754-64. doi: 10.1128/IAI.00917-15. Infect Immun. 2015. PMID: 26712207 Free PMC article.
References
-
- Akbar S, Kang C M, Gaidenko T A, Price C W. Modulator protein RsbR regulates environmental signalling in the general stress pathway of Bacillus subtilis. Mol Microbiol. 1997;24:567–578. - PubMed
-
- Altschul S F, Koonin E V. Iterated profile searches with PSI-BLAST—a tool for discovery in protein databases. Trends Biochem Sci. 1998;23:444–447. - PubMed
-
- Amin U S, Lash T D, Wilkinson B J. Proline betaine is a highly effective osmoprotectant for Staphylococcus aureus. Arch Microbiol. 1995;163:138–142. - PubMed
-
- Andersson S G, Zomorodipour A, Andersson J O, Sicheritz-Ponten T, Alsmark U C, Podowski R M, Naslund A K, Eriksson A S, Winkler H H, Kurland C G. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
