

Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins
- PMID: 10684945
- PMCID: PMC111042
- DOI: 10.1093/nar/28.6.1481
Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins
Abstract
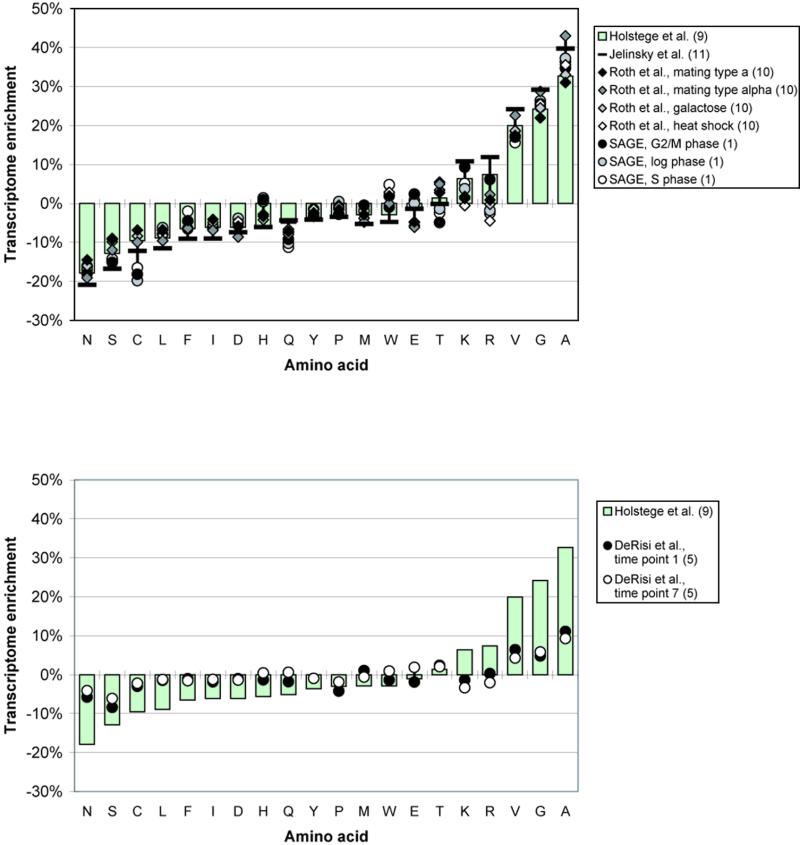
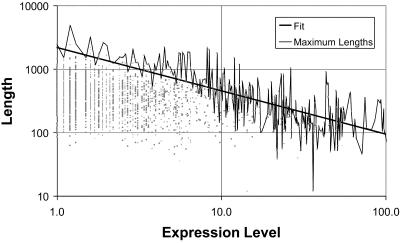
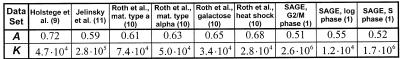
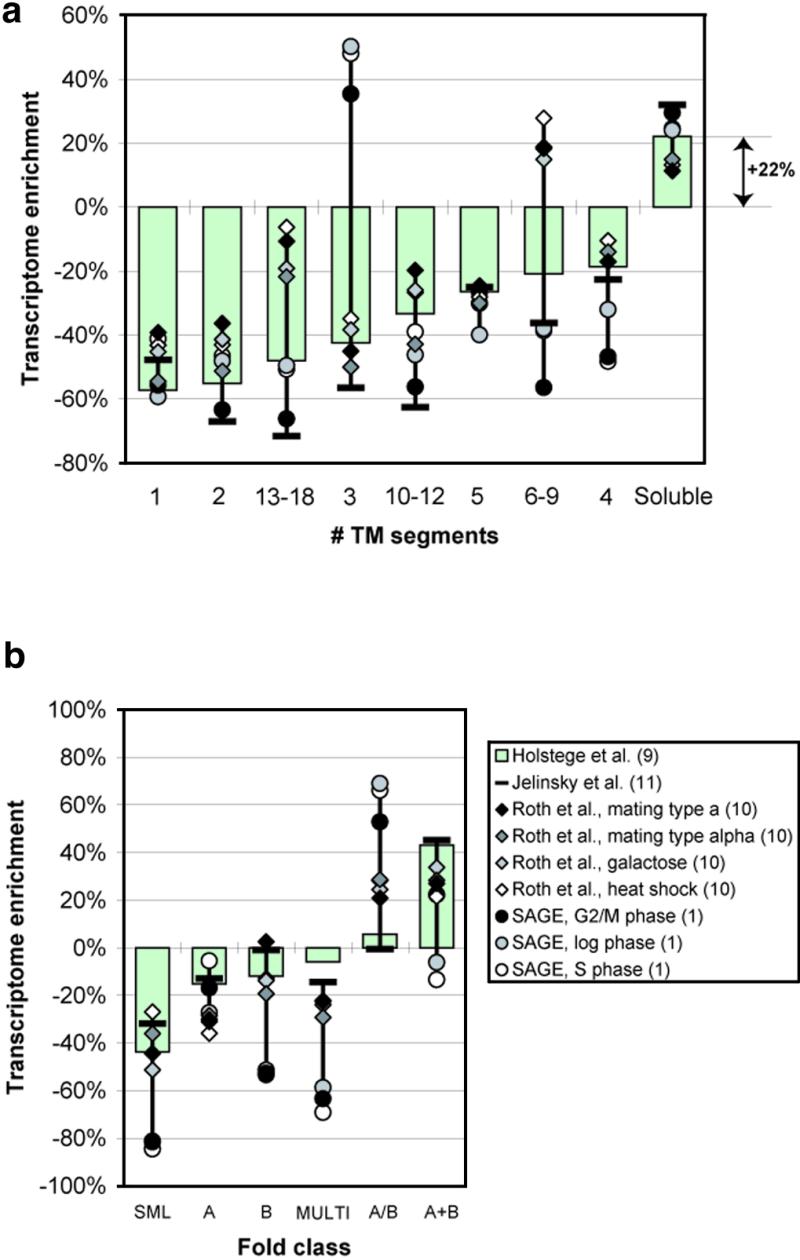
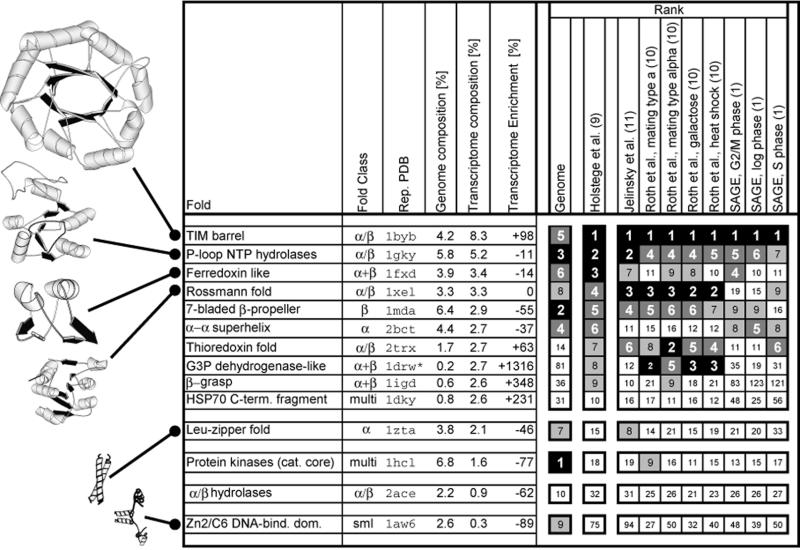
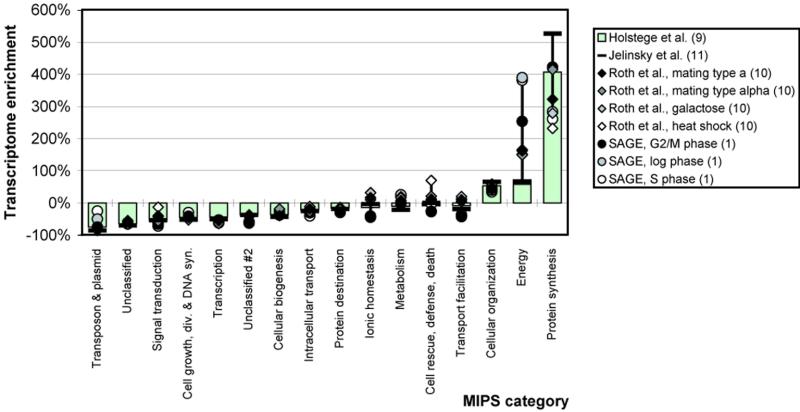
We analyzed 10 genome expression data sets by large-scale cross-referencing against broad structural and functional categories. The data sets, generated by different techniques (e.g. SAGE and gene chips), provide various representations of the yeast transcriptome (the set of all yeast genes, weighted by transcript abundance). Our analysis enabled us to determine features more prevalent in the transcriptome than the genome: i.e. those that are common to highly expressed proteins. Starting with simplest categories, we find that, relative to the genome, the transcriptome is enriched in Ala and Gly and depleted in Asn and very long proteins. We find, furthermore, that protein length and maximum expression level have a roughly inverse relationship. To relate expression level and protein structure, we assigned transmembrane helices and known folds (using PSI-blast) to each protein in the genome; this allowed us to determine that the transcriptome is enriched in mixed alpha-beta structures and depleted in membrane proteins relative to the genome. In particular, some enzymatic folds, such as the TIM barrel and the G3P dehydrogenase fold, are much more prevalent in the transcriptome than the genome, whereas others, such as the protein-kinase and leucine-zipper folds, are depleted. The TIM barrel, in fact, is overwhelmingly the 'top fold' in the transcriptome, while it only ranks fifth in the genome. The most highly enriched functional categories in the transcriptome (based on the MIPS system) are energy production and protein synthesis, while categories such as transcription, transport and signaling are depleted. Furthermore, for a given functional category, transcriptome enrichment varies quite substantially between the different expression data sets, with a variation an order of magnitude larger than for the other categories cross-referenced (e.g. amino acids). One can readily see how the enrichment and depletion of the various functional categories relates directly to that of particular folds.
Figures
References
-
- Velculescu V.E., Zhang,L., Zhou,W., Vogelstein,J., Basrai,M.A., Bassett,D.E.,Jr, Hieter,P., Vogelstein,B. and Kinzler,K.W. (1997) Cell, 88, 243–251. - PubMed
-
- Goffeau A., Barrell,B.G., Bussey,H., Davis,R.W., Dujon,B., Feldmann,H., Galibert,F., Hoheisel,J.D., Jacq,C., Johnston,M., Louis,E.J., Mewes,H.W., Murakami,Y., Philippsen,P., Tettelin,H. and Oliver,S.G. (1996) Science, 274, 546, 563–567. - PubMed
-
- Schena M., Shalon,D., Davis,R.W. and Brown,P.O. (1995) Science, 270, 467–470. - PubMed
-
- Shalon D., Smith,S.J. and Brown,P.O. (1996) Genome Res., 6, 639–645. - PubMed
-
- DeRisi J.L., Iyer,V.R. and Brown,P.O. (1997) Science, 278, 680–686. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
