

Selecting protein targets for structural genomics of Pyrobaculum aerophilum: validating automated fold assignment methods by using binary hypothesis testing
- PMID: 10706641
- PMCID: PMC15949
- DOI: 10.1073/pnas.050589297
Selecting protein targets for structural genomics of Pyrobaculum aerophilum: validating automated fold assignment methods by using binary hypothesis testing
Abstract
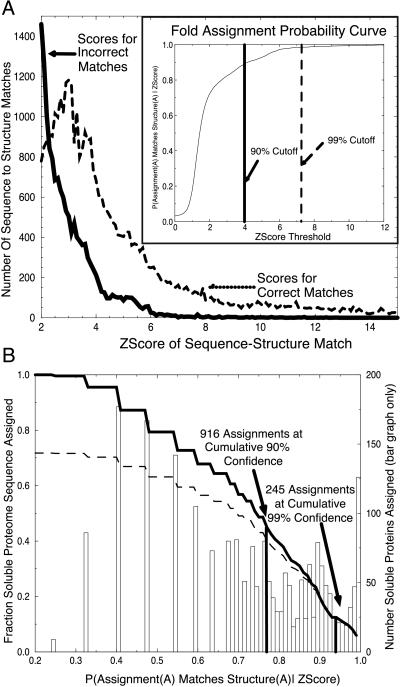
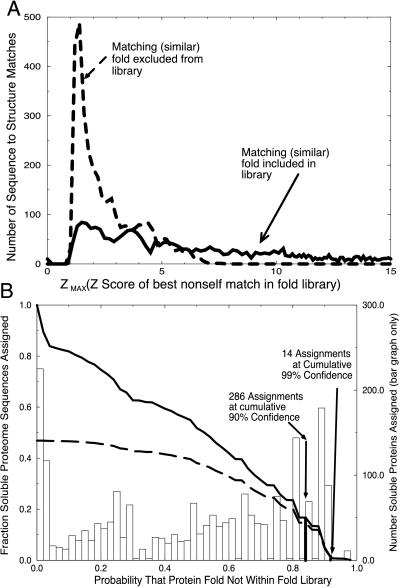
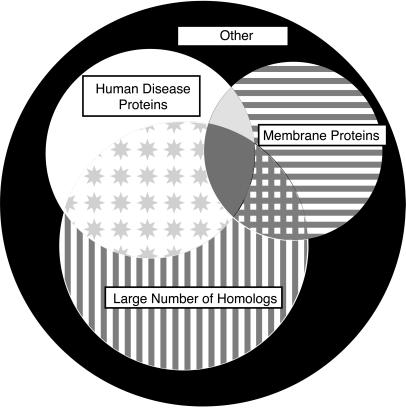
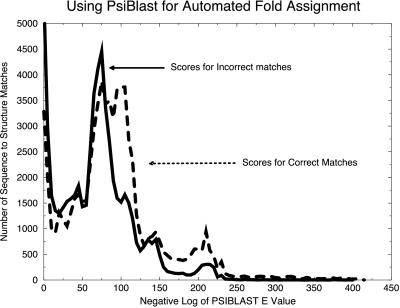
Three-dimensional protein folds were assigned to all ORFs of the recently sequenced genome of the hyperthermophilic archaeon Pyrobaculum aerophilum. Binary hypothesis testing was used to estimate a confidence level for each assignment. A separate test was conducted to assign a probability for whether each sequence has a novel fold-i.e., one that is not yet represented in the experimental database of known structures. Of the 2,130 predicted nontransmembrane proteins in this organism, 916 matched a fold at a cumulative 90% confidence level, and 245 could be assigned at a 99% confidence level. Likewise, 286 proteins were predicted to have a previously unobserved fold with a 90% confidence level, and 14 at a 99% confidence level. These statistically based tools are combined with homology searches against the Online Mendelian Inheritance in Man (OMIM) human genetics database and other protein databases for the selection of attractive targets for crystallographic or NMR structure determination. Results of these studies have been collated and placed at http://www.doe-mbi.ucla.edu/people/parag/P A_HOME/, the University of California, Los Angeles-Department of Energy Pyrobaculum aerophilum web site.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
