

Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain
- PMID: 10712197
- PMCID: PMC1288164
- DOI: 10.1086/302809
Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain
Abstract
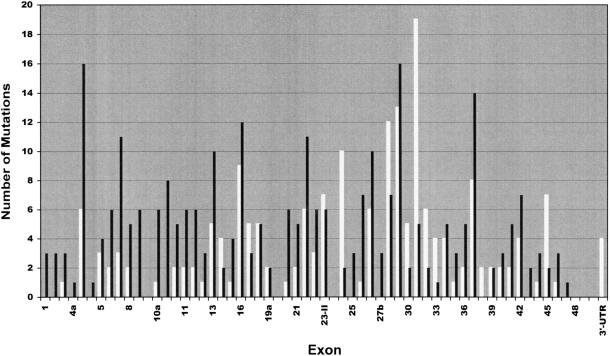
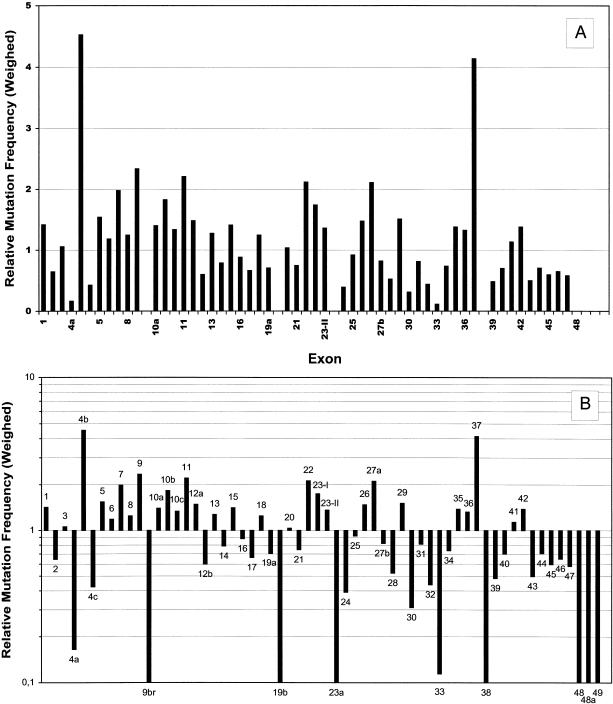
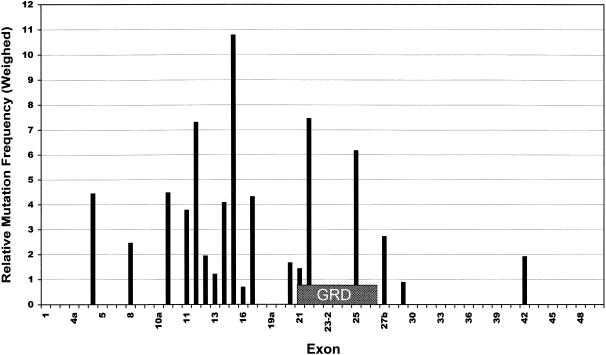
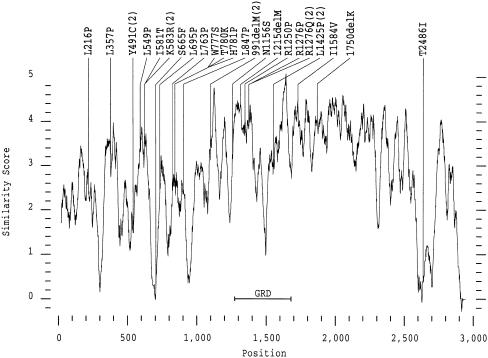
More than 500 unrelated patients with neurofibromatosis type 1 (NF1) were screened for mutations in the NF1 gene. For each patient, the whole coding sequence and all splice sites were studied for aberrations, either by the protein truncation test (PTT), temperature-gradient gel electrophoresis (TGGE) of genomic PCR products, or, most often, by direct genomic sequencing (DGS) of all individual exons. A total of 301 sequence variants, including 278 bona fide pathogenic mutations, were identified. As many as 216 or 183 of the genuine mutations, comprising 179 or 161 different ones, can be considered novel when compared to the recent findings of Upadhyaya and Cooper, or to the NNFF mutation database. Mutation-detection efficiencies of the various screening methods were similar: 47.1% for PTT, 53.7% for TGGE, and 54.9% for DGS. Some 224 mutations (80.2%) yielded directly or indirectly premature termination codons. These mutations showed even distribution over the whole gene from exon 1 to exon 47. Of all sequence variants determined in our study, <20% represent C-->T or G-->A transitions within a CpG dinucleotide, and only six different mutations also occur in NF1 pseudogenes, with five being typical C-->T transitions in a CpG. Thus, neither frequent deamination of 5-methylcytosines nor interchromosomal gene conversion may account for the high mutation rate of the NF1 gene. As opposed to the truncating mutations, the 28 (10.1%) missense or single-amino-acid-deletion mutations identified clustered in two distinct regions, the GAP-related domain (GRD) and an upstream gene segment comprising exons 11-17. The latter forms a so-called cysteine/serine-rich domain with three cysteine pairs suggestive of ATP binding, as well as three potential cAMP-dependent protein kinase (PKA) recognition sites obviously phosphorylated by PKA. Coincidence of mutated amino acids and those conserved between human and Drosophila strongly suggest significant functional relevance of this region, with major roles played by exons 12a and 15 and part of exon 16.
Figures

References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for human and Drosophila neurofibromin protein sequences [accession numbers AAA59925 and AAB58976, respectively] and human NF1 pseudogene sequences [accession numbers are listed in ])
-
- International NF1 Genetic Analysis Consortium, http://www.nf.org/nf1gene/nf1gene.home.html (for unpublished NF1 mutations)
-
- Messiaen L, Callens T, Mortier G, van Roy N, Speleman F, de Pape A (1998) Identification of mutations in the NF1 gene, including 3 different nonsense mutations and 1 missense mutation disrupting normal RNA splicing. (Abstract) http://nf.org/md1aspe1.htm
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for NF1 [162200])
References
-
- Abernathy CR, Rasmussen SA, Stalker HJ, Zori R, Driscoll DJ, Williams CA, Kousseff BG, et al (1997) NF1 mutation analysis using a combined heteroduplex/SSCP approach. Hum Mutat 9:548–554 - PubMed
-
- Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A (1997) Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat Struct Biol 4:686–689 - PubMed
-
- Ainsworth PJ, Chakraborty PK, Weksberg R (1997) Example of somatic mosaicism in a series of de novo neurofibromatosis type 1 cases due to a maternally derived deletion. Hum Mutat 9:452–457 - PubMed
-
- Andrews JD, Mancini DN, Singh SM, Rodenhiser DI (1996) Site and sequence specific DNA methylation in the neurofibromatosis (NF1) gene includes C5839T: the site of the recurrent substitution mutation in exon 31. Hum Mol Genet 5:503–507 - PubMed
-
- Antonarakis SE, Kazazian HH, Tuddenham EG (1995) Molecular etiology of factor VIII deficiency in hemophilia A. Hum Mutat 5:1–22 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
