

COL2A1 exon 2 mutations: relevance to the Stickler and Wagner syndromes
- PMID: 10729292
- PMCID: PMC1723423
- DOI: 10.1136/bjo.84.4.364
COL2A1 exon 2 mutations: relevance to the Stickler and Wagner syndromes
Abstract
Aims: To compare the clinical and molecular genetic features of two phenotypically distinct subgroups of families with type 1 Stickler syndrome.
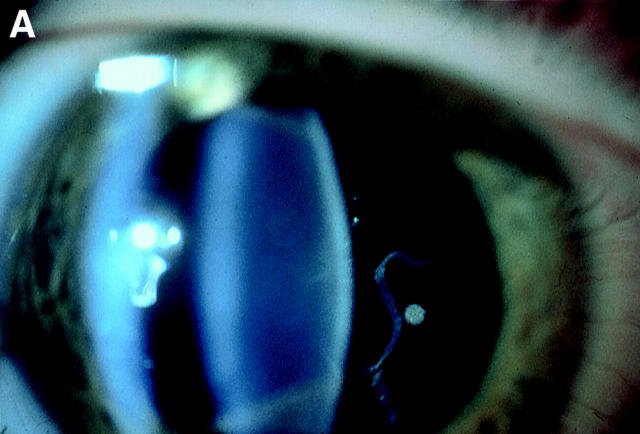
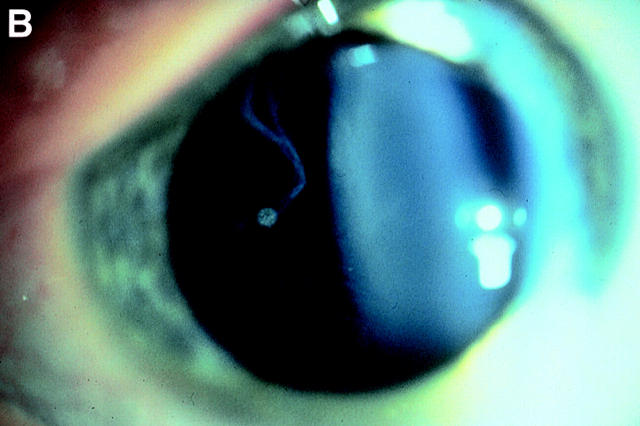
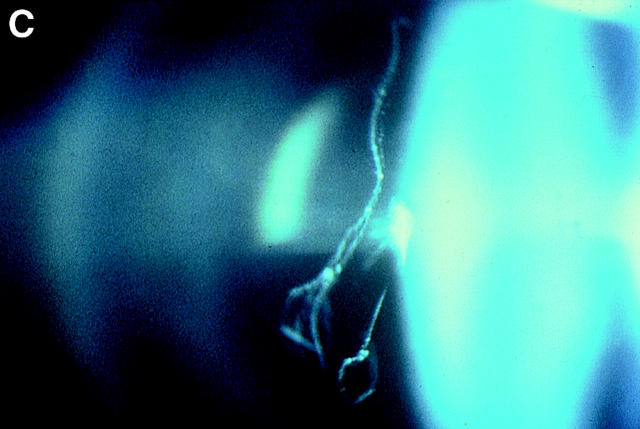
Background: Stickler syndrome (hereditary arthro-ophthalmopathy, McKusick Nos 108300 and 184840) is a dominantly inherited disorder of collagen connective tissue, resulting in an abnormal vitreous, myopia, and a variable degree of orofacial abnormality, deafness, and arthropathy. Stickler syndrome is the commonest inherited cause of rhegmatogenous retinal detachment in childhood with a risk of giant retinal tear (GRT) which is commonly bilateral and a frequent cause of blindness.
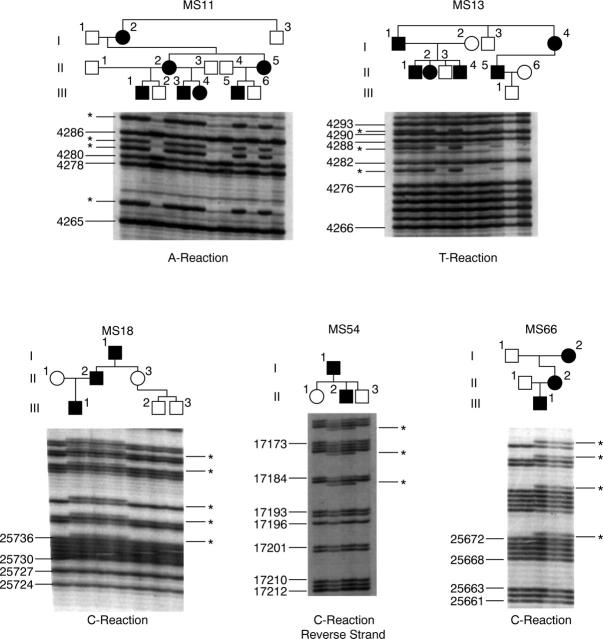
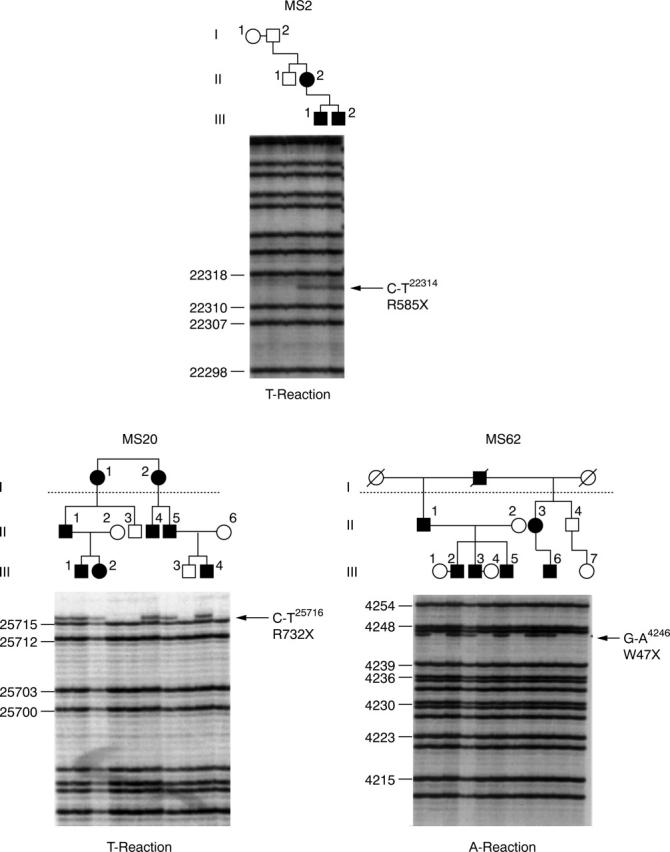
Method: Pedigrees were identified from the vitreoretinal service database and subclassified according to vitreoretinal phenotype. Ophthalmic, skeletal, auditory, and orofacial features were assessed. Linkage analysis was carried out with markers for the candidate genes COL2A1, COL11A1, and COL11A2. The COL2A1 gene was amplified as five overlapping PCR products. Direct sequencing of individual exons identified mutations.
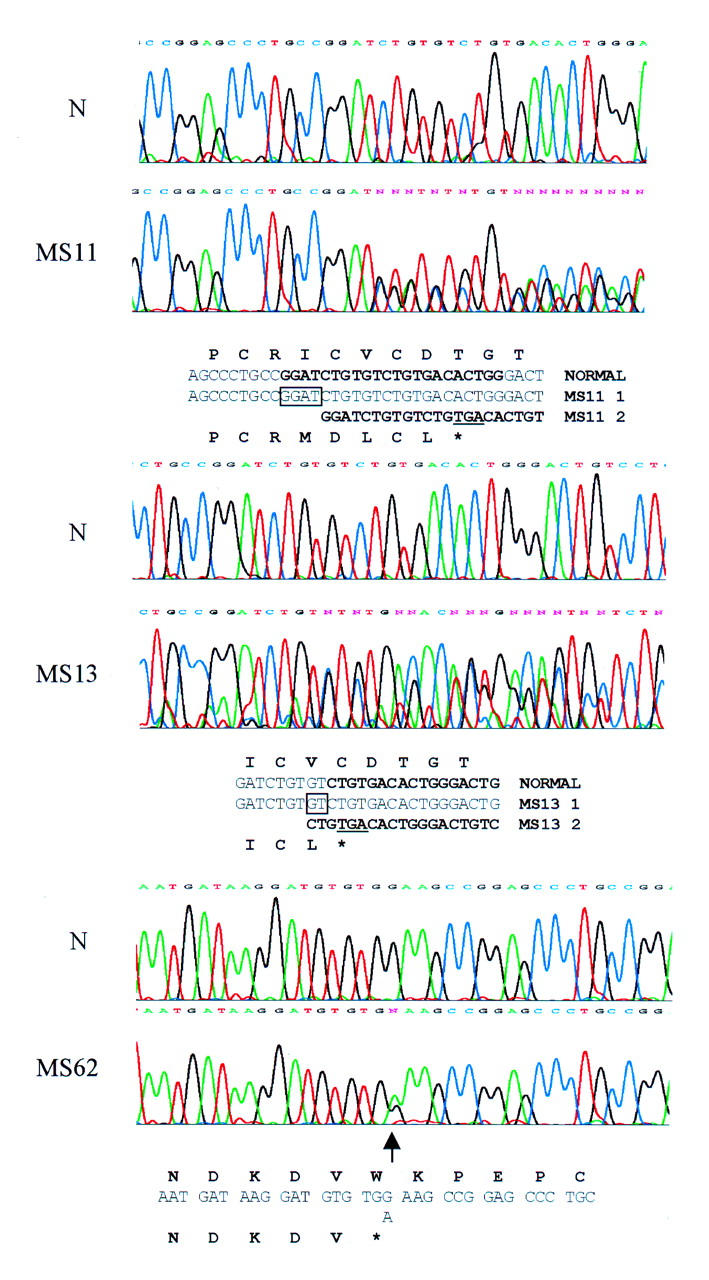
Results: Eight families exhibiting the type 1 vitreous phenotype were studied. Seven were consistent for linkage to COL2A1, with lod scores ranging from 2.1 to 0.3. In most instances linkage to COL11A1 and COL11A2 could be excluded. One family was analysed without prior linkage analysis. Three of the families exhibited a predominantly ocular phenotype with minimal or absent systemic involvement and were found to have mutations in exon 2 of COL2A1. Five other pedigrees with an identical ocular phenotype plus orofacial, auditory, and articular involvement had mutations in others regions of the COL2A1 gene. None of the pedigrees exhibited the characteristic lenticular, retinal pigment epithelial, or choroidal changes seen in Wagner syndrome.
Conclusions: These data confirm that type 1 Stickler syndrome is caused by mutations in the gene encoding type II collagen (COL2A1). In addition, data are submitted showing that mutations involving exon 2 of COL2A1 are characterised by a predominantly ocular variant of this disorder, consistent with the major form of type II procollagen in non-ocular tissues having exon 2 spliced out. Such patients are all at high risk of retinal detachment. This has important implications for counselling patients with regard to the development of systemic complications. It also emphasises the importance and reliability of the ophthalmic examination in the differential diagnosis of this predominantly ocular form of Stickler syndrome from Wagner's vitreoretinopathy.
Figures
Comment in
-
A frame shift mutation in a tissue-specific alternatively spliced exon of collagen 2A1 in Wagner's vitreoretinal degeneration.Am J Ophthalmol. 2002 Sep;134(3):473; author reply 473-4. doi: 10.1016/s0002-9394(02)01635-5. Am J Ophthalmol. 2002. PMID: 12208278 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous