

Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C
- PMID: 10739751
- PMCID: PMC1288186
- DOI: 10.1086/302836
Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C
Erratum in
- Am J Hum Genet 2000 Sep;67(3):775
Abstract
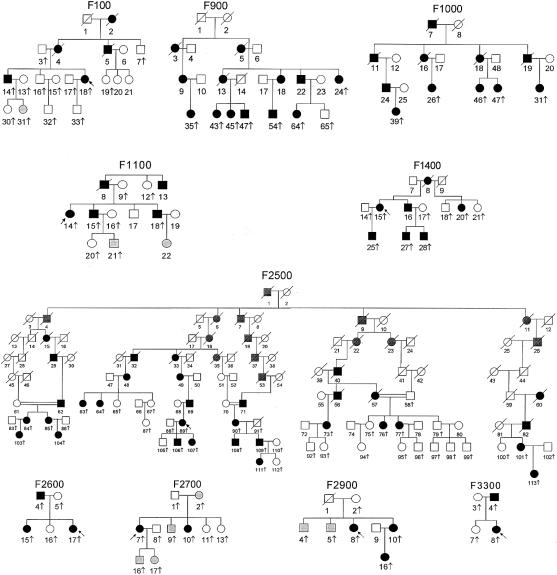
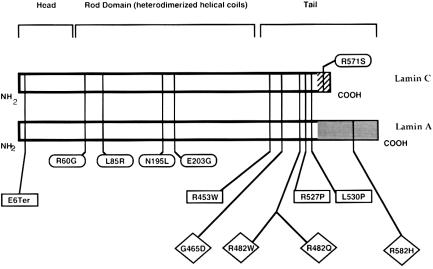
Familial partial lipodystrophy (FPLD), Dunnigan variety, is an autosomal dominant disorder characterized by marked loss of subcutaneous adipose tissue from the extremities and trunk but by excess fat deposition in the head and neck. The disease is frequently associated with profound insulin resistance, dyslipidemia, and diabetes. We have localized a gene for FPLD to chromosome 1q21-q23, and it has recently been proposed that nuclear lamin A/C is altered in FPLD, on the basis of a novel missense mutation (R482Q) in five Canadian probands. This gene had previously been shown to be altered in autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD-AD) and in dilated cardiomyopathy and conduction-system disease. We examined 15 families with FPLD for mutations in lamin A/C. Five families harbored the R482Q alteration that segregated with the disease phenotype. Seven families harbored an R482W alteration, and one family harbored a G465D alteration. All these mutations lie within exon 8 of the lamin A/C gene-an exon that has also been shown to harbor different missense mutations that are responsible for EDMD-AD. Mutations could not be detected in lamin A/C in one FPLD family in which there was linkage to chromosome 1q21-q23. One family with atypical FPLD harbored an R582H alteration in exon 11 of lamin A. This exon does not comprise part of the lamin C coding region. All mutations in FPLD affect the globular C-terminal domain of the lamin A/C protein. In contrast, mutations responsible for dilated cardiomyopathy and conduction-system disease are observed in the rod domain of the protein. The FPLD mutations R482Q and R482W occurred on different haplotypes, indicating that they are likely to have arisen more than once.
Figures

References
Electronic-Database Information
-
- CEPH, http://www.cephb.fr/
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for human nuclear lamin A and nuclear lamin C gene, exon 1 [accession number L12399]; human nuclear lamin A and nuclear lamin C gene, exon 2 [accession number L12400]; and human nuclear lamin A and nuclear lamin C gene, exons 3–12, and complete alternative mRNAs [accession number L12401])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for FPLD [MIM 151660])
References
-
- Anderson JL, Khan M, David WS, Mahdavi Z, Nuttall FQ, Krech E, West SG, et al (1999) Confirmation of linkage of hereditary partial lipodystrophy to chromosome 1q21-22. Am J Med Genet 82:161–165 - PubMed
-
- Bennett LB, Roach SE, Bowcock AM (2000) A locus for paroxysmal kinesigenic dyskinesia maps to human chromosome 16. Neurology 54:125–130 - PubMed
-
- Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, et al (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 21:285–288 - PubMed
-
- Cao H, Hegele RA (2000) Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 9:109–112 - PubMed
-
- Doskocil J, Sorm F (1962) Distribution of 5-methylcytosine in pyrimidine sequences of deoxyribonucleic acids. Biochim Biophys Acta 55:953–959 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
