

Dual control of muscle cell survival by distinct growth factor-regulated signaling pathways
- PMID: 10757809
- PMCID: PMC85619
- DOI: 10.1128/MCB.20.9.3256-3265.2000
Dual control of muscle cell survival by distinct growth factor-regulated signaling pathways
Abstract
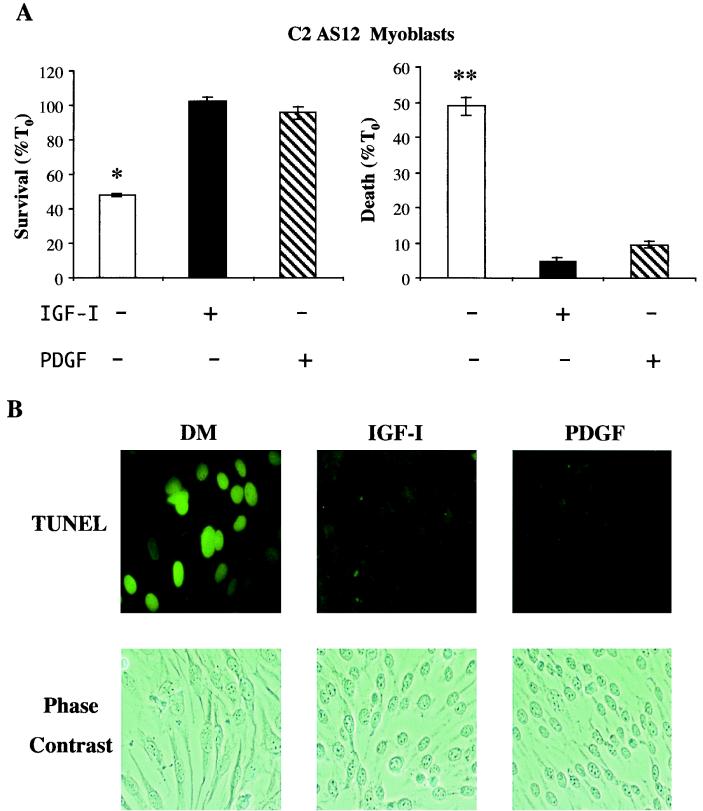
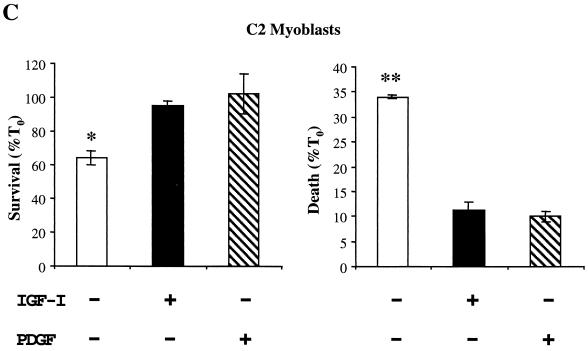
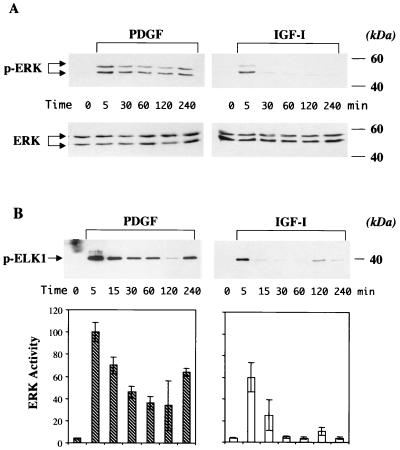
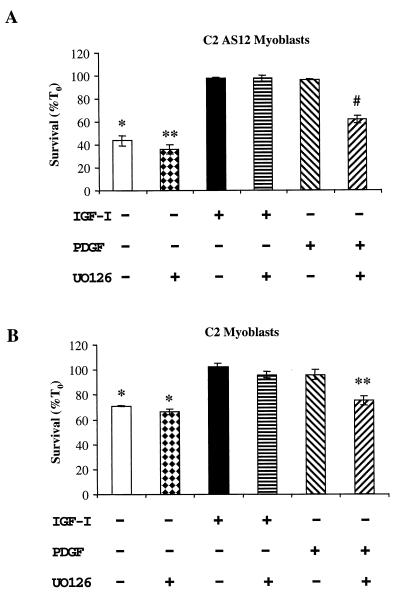
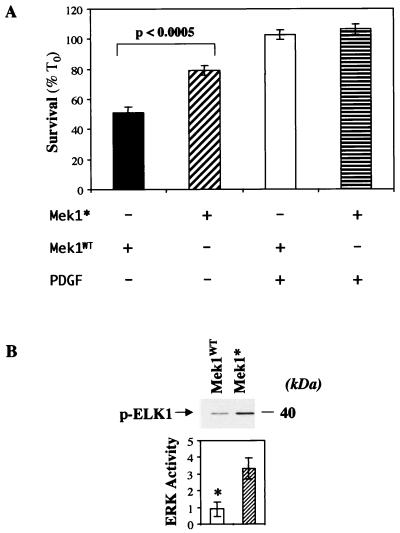
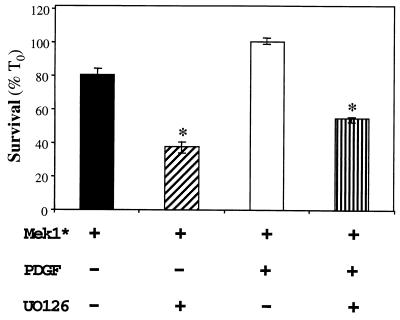
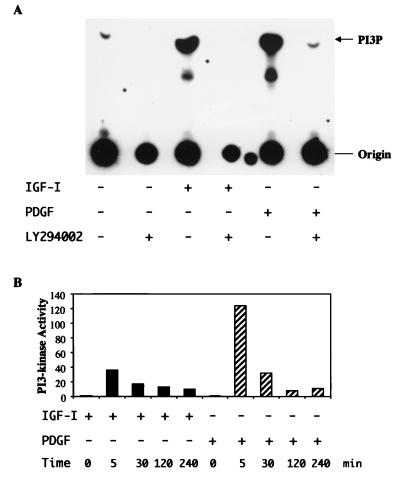
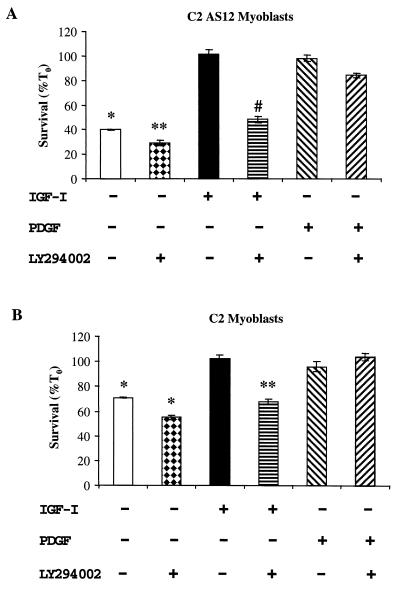
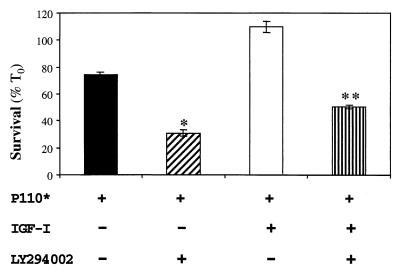
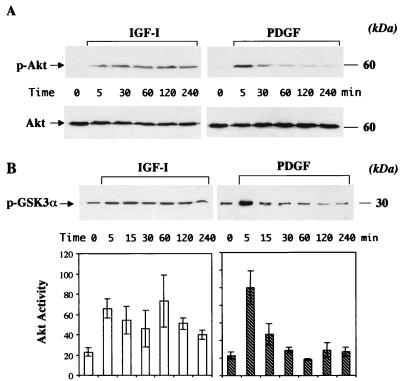
In addition to their ability to stimulate cell proliferation, polypeptide growth factors are able to maintain cell survival under conditions that otherwise lead to apoptotic death. Growth factors control cell viability through regulation of critical intracellular signal transduction pathways. We previously characterized C2 muscle cell lines that lacked endogenous expression of insulin-like growth factor II (IGF-II). These cells did not differentiate but underwent apoptotic death in low-serum differentiation medium. Death could be prevented by IGF analogues that activated the IGF-I receptor or by unrelated growth factors such as platelet-derived growth factor BB (PDGF-BB). Here we analyze the signaling pathways involved in growth factor-mediated myoblast survival. PDGF treatment caused sustained activation of extracellular-regulated kinases 1 and 2 (ERK1 and -2), while IGF-I only transiently induced these enzymes. Transient transfection of a constitutively active Mek1, a specific upstream activator of ERKs, maintained myoblast viability in the absence of growth factors, while inhibition of Mek1 by the drug UO126 blocked PDGF-mediated but not IGF-stimulated survival. Although both growth factors activated phosphatidylinositol 3-kinase (PI3-kinase) to similar extents, only IGF-I treatment led to sustained stimulation of its downstream kinase, Akt. Transient transfection of a constitutively active PI3-kinase or an inducible Akt promoted myoblast viability in the absence of growth factors, while inhibition of PI3-kinase activity by the drug LY294002 selectively blocked IGF- but not PDGF-mediated muscle cell survival. In aggregate, these observations demonstrate that distinct growth factor-regulated signaling pathways independently control myoblast survival. Since IGF action also stimulates muscle differentiation, these results suggest a means to regulate myogenesis through selective manipulation of different signal transduction pathways.
Figures


Similar articles
-
Changes in the balance of phosphoinositide 3-kinase/protein kinase B (Akt) and the mitogen-activated protein kinases (ERK/p38MAPK) determine a phenotype of visceral and vascular smooth muscle cells.J Cell Biol. 1999 May 17;145(4):727-40. doi: 10.1083/jcb.145.4.727. J Cell Biol. 1999. PMID: 10330402 Free PMC article.
-
Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21.Mol Cell Biol. 2000 Dec;20(23):8983-95. doi: 10.1128/MCB.20.23.8983-8995.2000. Mol Cell Biol. 2000. PMID: 11073997 Free PMC article.
-
Platelet-derived growth factor-BB, insulin-like growth factor-I, and phorbol ester activate different signaling pathways for stimulation of vascular smooth muscle cell migration.Exp Cell Res. 1998 Aug 1;242(2):548-60. doi: 10.1006/excr.1998.4138. Exp Cell Res. 1998. PMID: 9683541
-
The signaling landscape of insulin-like growth factor 1.J Biol Chem. 2025 Jan;301(1):108047. doi: 10.1016/j.jbc.2024.108047. Epub 2024 Dec 3. J Biol Chem. 2025. PMID: 39638246 Free PMC article. Review.
-
TRPV2: A Calcium-Permeable Cation Channel Regulated by Insulin-Like Growth Factors.In: Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007. Chapter 7. In: Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis; 2007. Chapter 7. PMID: 21204502 Free Books & Documents. Review.
Cited by
-
Phosphatidylinositol 3-kinase-binding protein, PI3KAP/XB130, is required for cAMP-induced amplification of IGF mitogenic activity in FRTL-5 thyroid cells.Mol Endocrinol. 2012 Jun;26(6):1043-55. doi: 10.1210/me.2011-1349. Epub 2012 Apr 11. Mol Endocrinol. 2012. PMID: 22496359 Free PMC article.
-
IGF-1-Mediated Survival from Induced Death of Human Primary Cultured Retinal Pigment Epithelial Cells Is Mediated by an Akt-Dependent Signaling Pathway.Mol Neurobiol. 2018 Mar;55(3):1915-1927. doi: 10.1007/s12035-017-0447-0. Epub 2017 Feb 25. Mol Neurobiol. 2018. PMID: 28238097
-
TIMP3: a physiological regulator of adult myogenesis.J Cell Sci. 2010 Sep 1;123(Pt 17):2914-21. doi: 10.1242/jcs.057620. Epub 2010 Aug 3. J Cell Sci. 2010. PMID: 20682640 Free PMC article.
-
Different Effects of Insulin-Like Growth Factor-1 and Insulin-Like Growth Factor-2 on Myogenic Differentiation of Human Mesenchymal Stem Cells.Stem Cells Int. 2017;2017:8286248. doi: 10.1155/2017/8286248. Epub 2017 Dec 14. Stem Cells Int. 2017. PMID: 29387091 Free PMC article.
-
Live cell imaging reveals marked variability in myoblast proliferation and fate.Skelet Muscle. 2013 May 2;3(1):10. doi: 10.1186/2044-5040-3-10. Skelet Muscle. 2013. PMID: 23638706 Free PMC article.
References
-
- Baserga R, Resnicoff M, D'Ambrosio C, Valentinis B. The role of the IGF-I receptor in apoptosis. Vitam Horm. 1997;53:65–98. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous