

Slow death of postnatal hippocampal neurons by GABA(A) receptor overactivation
- PMID: 10777778
- PMCID: PMC6773103
- DOI: 10.1523/JNEUROSCI.20-09-03147.2000
Slow death of postnatal hippocampal neurons by GABA(A) receptor overactivation
Abstract
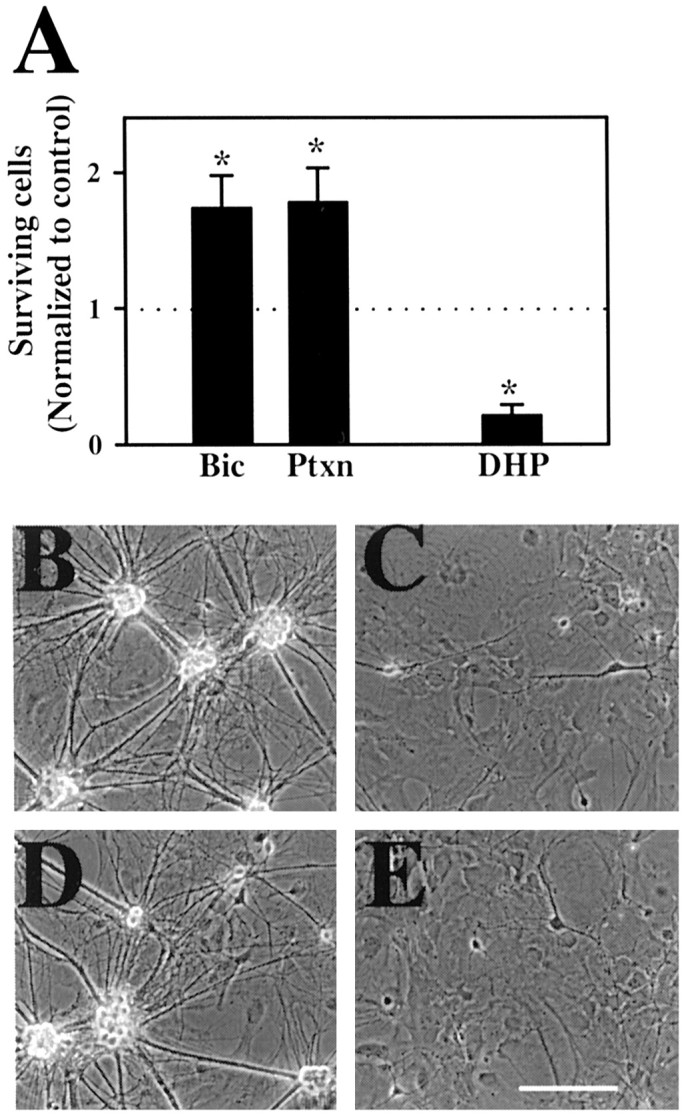
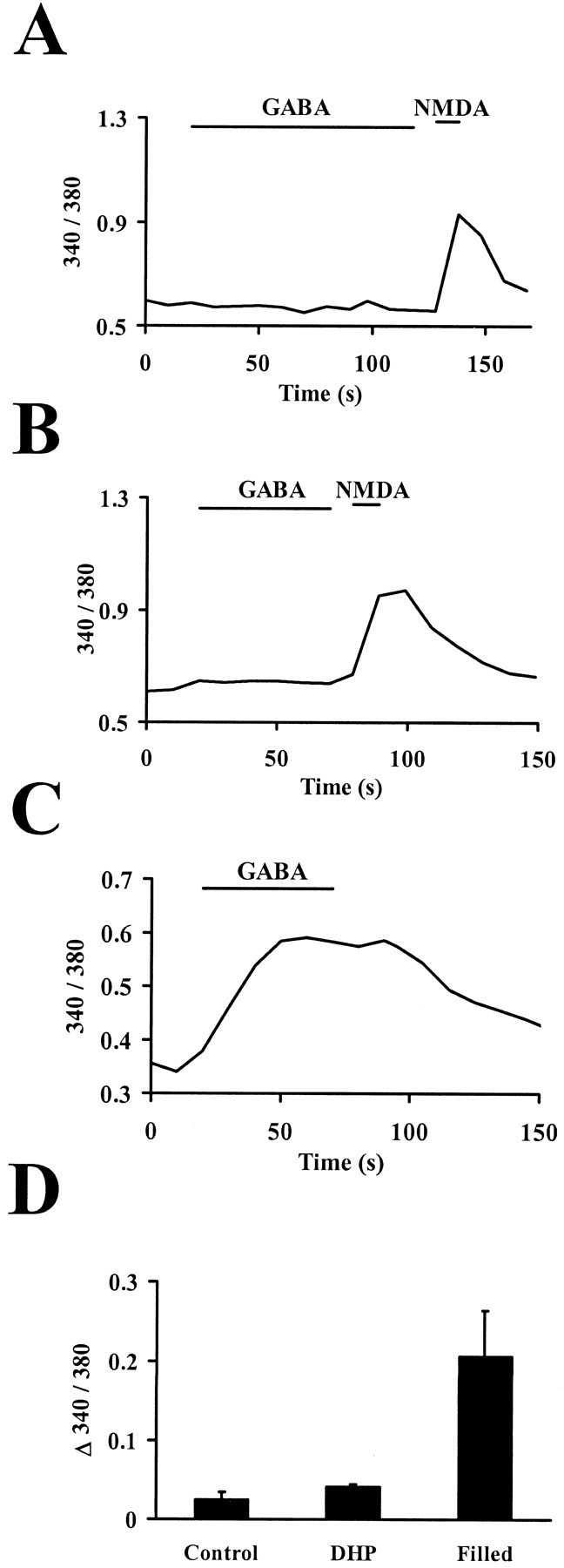
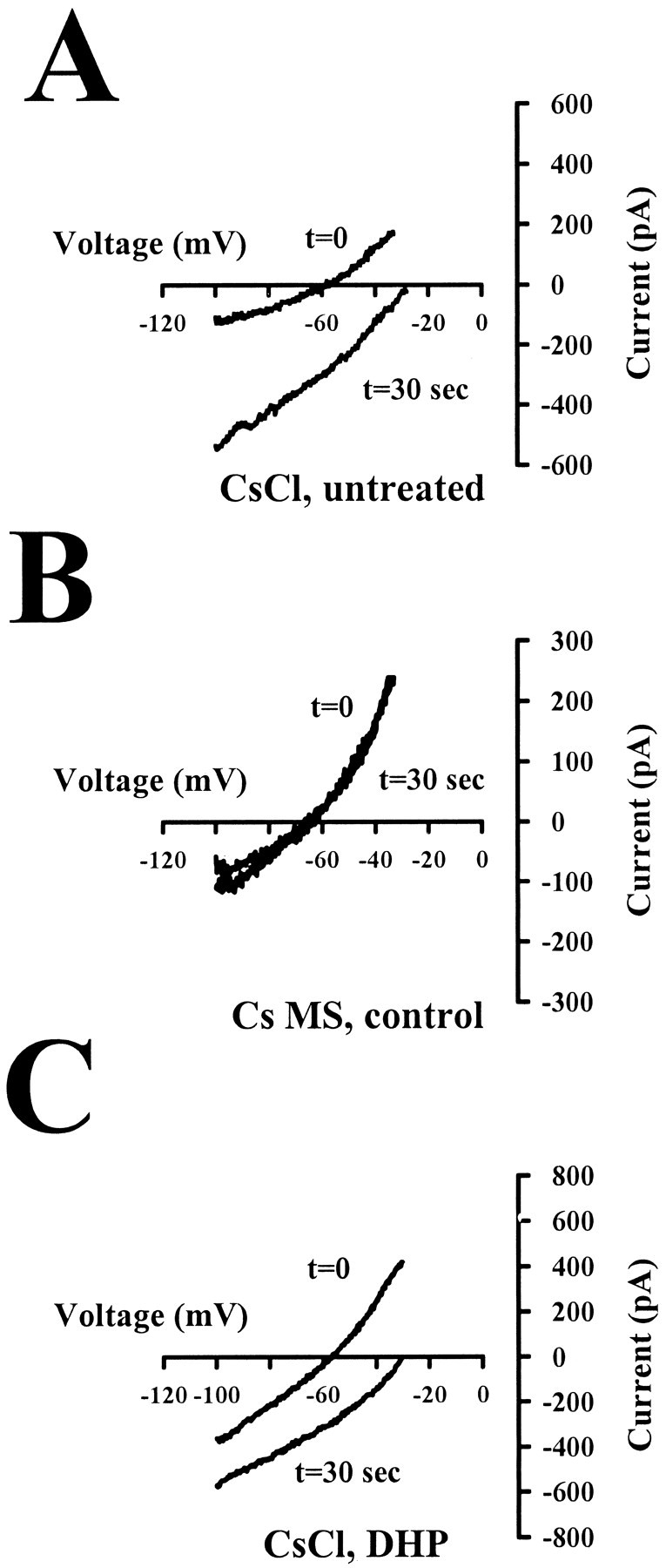
Neurotransmitters can have both toxic and trophic functions in addition to their role in neural signaling. Surprisingly, chronic blockade of GABA(A) receptor activity for 5-8 d in vitro enhanced survival of hippocampal neurons, suggesting that GABA(A) receptor overactivation may be neurotoxic. Potentiating GABA(A) receptor activity by chronic treatment with the endogenous neurosteroid (3alpha,5alpha)-3-hydroxypregnan-20-one caused massive cell loss over 1 week in culture. Other potentiators of GABA(A) receptors, including benzodiazepines, mimicked the cell loss, suggesting that potentiating endogenous GABA activity is sufficient to produce neuronal death. Neurosteroid-treated neurons had lower resting intracellular calcium levels than control cells and produced smaller calcium rises in response to depolarizing challenges. Manipulating intracellular calcium levels with chronic elevated extracellular potassium or with the calcium channel agonist Bay K 8644 protected neurons. The results may have implications for the mechanisms of programmed cell death in the developing CNS as well as implications for the long-term consequences of chronic GABAmimetic drug use during development.
Figures






References
-
- Ankarcrona M. Glutamate induced cell death: apoptosis or necrosis? Prog Brain Res. 1998;116:265–272. - PubMed
-
- Castoldi AF, Barni S, Randine G, Costa LG, Manzo L. Ethanol selectively interferes with the trophic action of NMDA and carbachol on cultured cerebellar granule neurons undergoing apoptosis. Brain Res Dev Brain Res. 1998;111:279–289. - PubMed
-
- Chen Q, Olney JW, Lukasiewicz PD, Almli T, Romano C. Ca2+-independent excitotoxic neurodegeneration in isolated retina, an intact neural net: a role for Cl− and inhibitory transmitters. Mol Pharmacol. 1998;53:564–572. - PubMed
-
- Cherubini E, Rovira C, Gaiarsa JL, Corradetti R, Ben Ari Y. GABA mediated excitation in immature rat CA3 hippocampal neurons. Int J Dev Neurosci. 1990;8:481–490. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources