

The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription
- PMID: 10823891
- PMCID: PMC18731
- DOI: 10.1073/pnas.100110097
The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription
Abstract
Huntington's Disease (HD) is caused by an expansion of a polyglutamine tract within the huntingtin (htt) protein. Pathogenesis in HD appears to include the cytoplasmic cleavage of htt and release of an amino-terminal fragment capable of nuclear localization. We have investigated potential consequences to nuclear function of a pathogenic amino-terminal region of htt (httex1p) including aggregation, protein-protein interactions, and transcription. httex1p was found to coaggregate with p53 in inclusions generated in cell culture and to interact with p53 in vitro and in cell culture. Expanded httex1p represses transcription of the p53-regulated promoters, p21(WAF1/CIP1) and MDR-1. httex1p was also found to interact in vitro with CREB-binding protein (CBP) and mSin3a, and CBP to localize to neuronal intranuclear inclusions in a transgenic mouse model of HD. These results raise the possibility that expanded repeat htt causes aberrant transcriptional regulation through its interaction with cellular transcription factors which may result in neuronal dysfunction and cell death in HD.
Figures
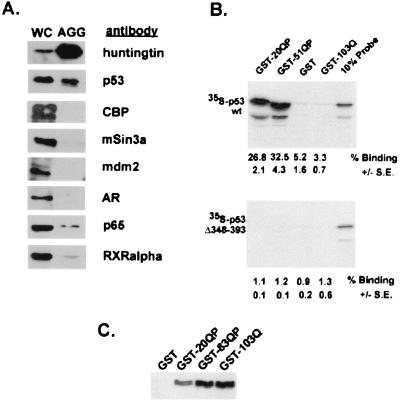
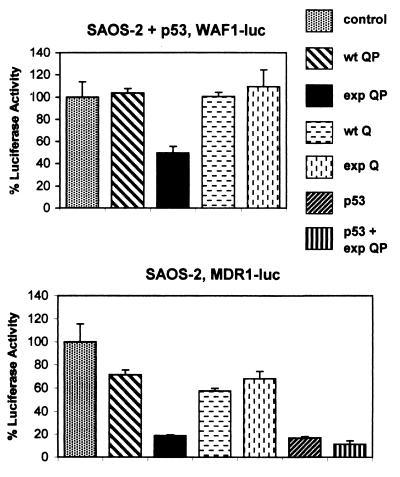
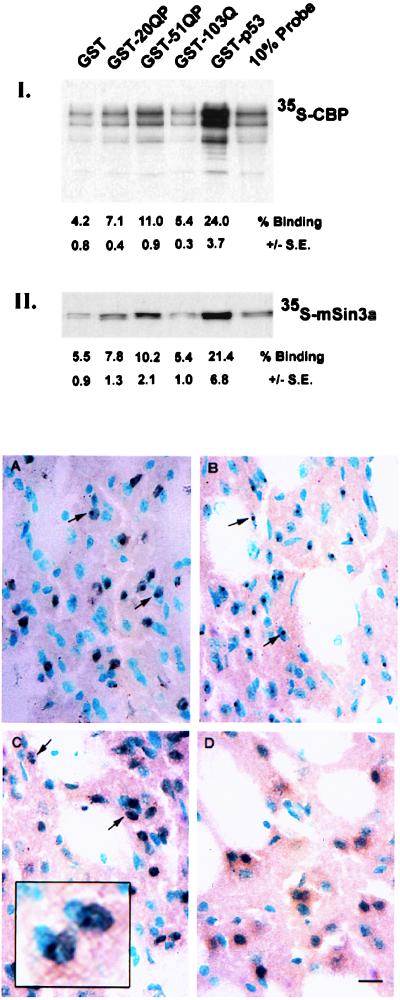
Comment in
-
Novel therapies in the search for a cure for Huntington's disease.Proc Natl Acad Sci U S A. 2001 Jan 2;98(1):3-4. doi: 10.1073/pnas.98.1.3. Proc Natl Acad Sci U S A. 2001. PMID: 11136240 Free PMC article. No abstract available.
References
-
- Harper P S, editor. Huntington's Disease. London: W. B. Saunders; 1991. pp. 141–178.
-
- The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. - PubMed
-
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Yound C, Martin E, Vonsattel J-P, Carraway R, Reeves S A, Boyce F M, Aronin N. Neuron. 1995;14:1075–1081. - PubMed
-
- Gusella J F, MacDonald M E. Semin Cell Biol. 1995;6:21–28. - PubMed
-
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies S W, Bates G P. Cell. 1996;87:493–506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
