

Modeling RNA folding paths with pseudoknots: application to hepatitis delta virus ribozyme
- PMID: 10823910
- PMCID: PMC18642
- DOI: 10.1073/pnas.110533697
Modeling RNA folding paths with pseudoknots: application to hepatitis delta virus ribozyme
Abstract
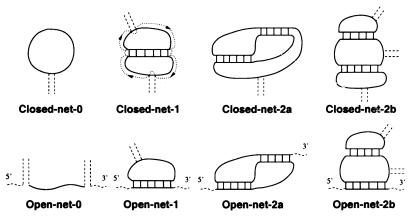
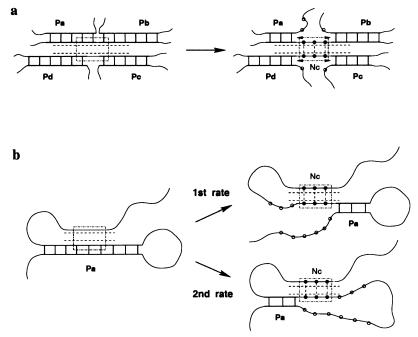
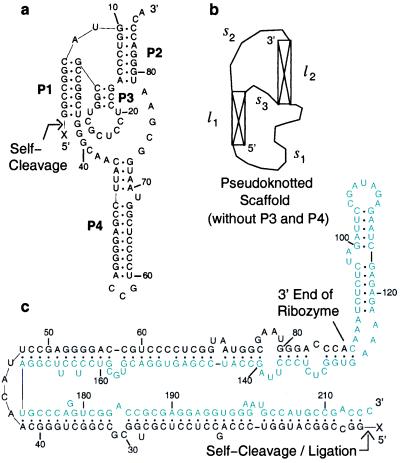
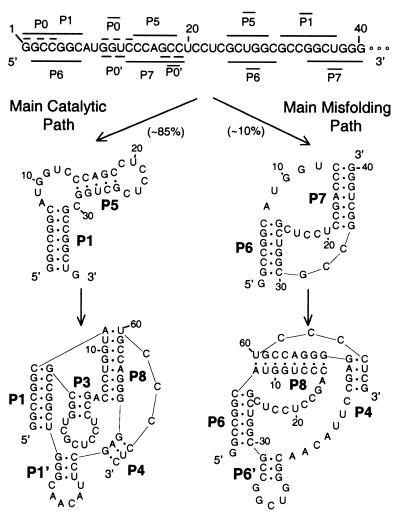
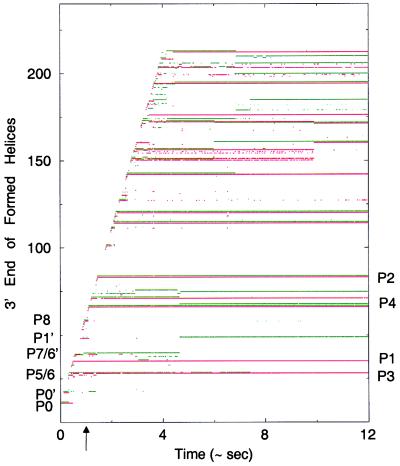
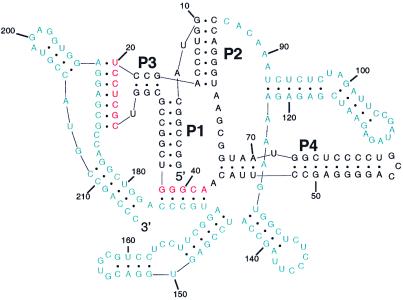
A quantitative understanding of nucleic acid hybridization is essential to many aspects of biotechnology, such as DNA microarrays, as well as to the structure and folding kinetics of RNA. However, predictions of nucleic acid secondary structures have long been impeded by the presence of helices interior to loops, so-called pseudoknots, which impose complex three-dimensional conformational constraints. In this paper we compute the pseudoknot free energies analytically in terms of known standard parameters, and we show how the results can be included in a kinetic Monte Carlo code to follow the succession of secondary structures during quenched or sequential folding. For the hepatitis delta virus ribozyme, we predict several nonnative stems on the folding path, characterize a kinetically trapped state, interpret several experimentally characterized mutations in terms of the folding path, and suggest how hybridization with other parts of the genome inactivates the newly formed ribozyme.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
