

DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints
- PMID: 10825202
- PMCID: PMC85806
- DOI: 10.1128/MCB.20.12.4393-4404.2000
DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints
Abstract
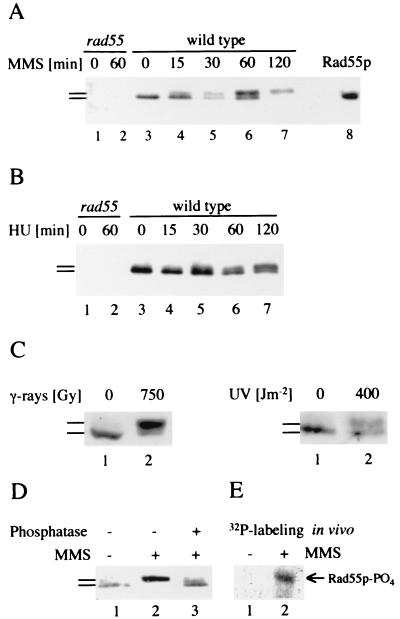
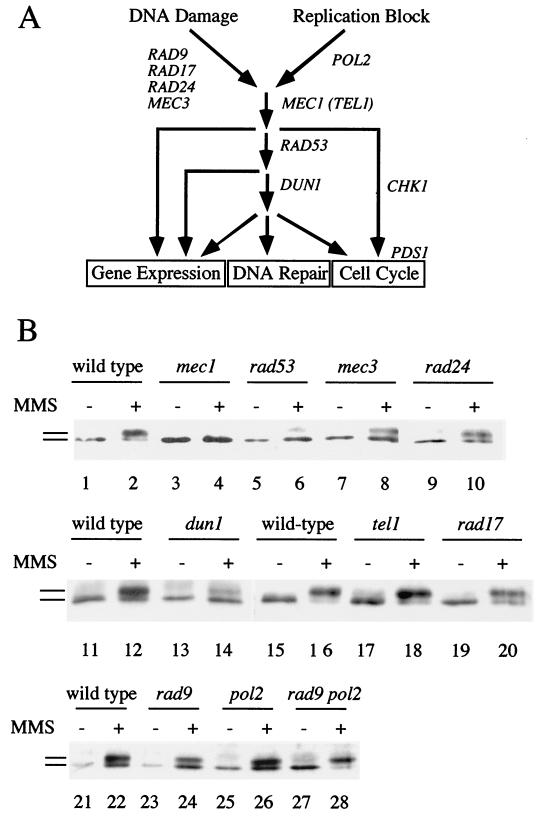
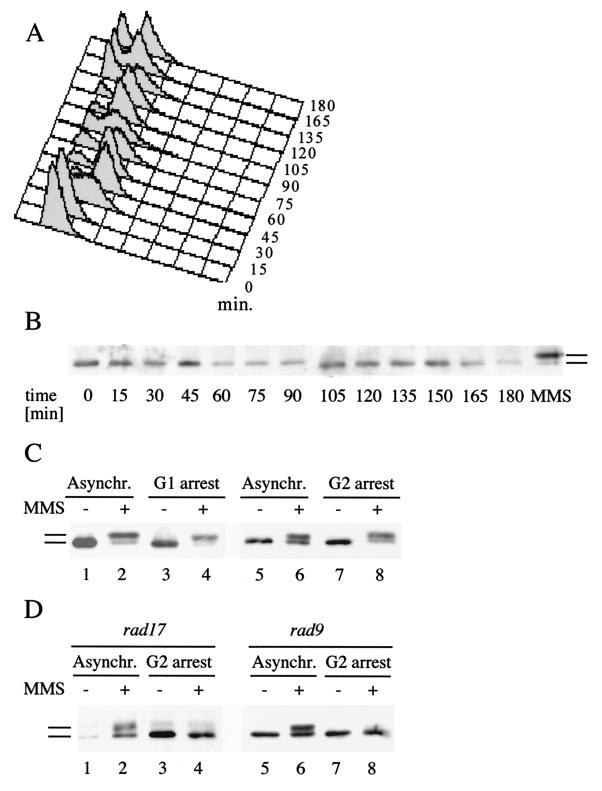
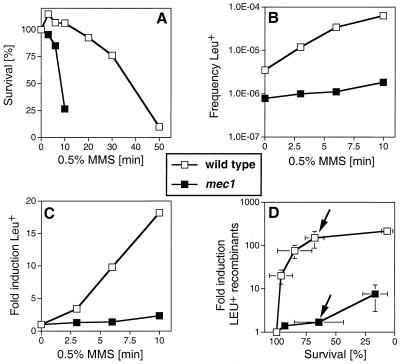
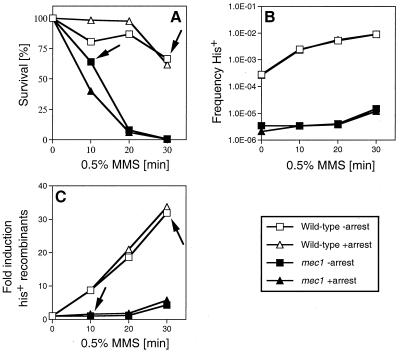
Checkpoints, which are integral to the cellular response to DNA damage, coordinate transient cell cycle arrest and the induced expression of DNA repair genes after genotoxic stress. DNA repair ensures cellular survival and genomic stability, utilizing a multipathway network. Here we report evidence that the two systems, DNA damage checkpoint control and DNA repair, are directly connected by demonstrating that the Rad55 double-strand break repair protein of the recombinational repair pathway is a terminal substrate of DNA damage and replication block checkpoints. Rad55p was specifically phosphorylated in response to DNA damage induced by the alkylating agent methyl methanesulfonate, dependent on an active DNA damage checkpoint. Rad55p modification was also observed after gamma ray and UV radiation. The rapid time course of phosphorylation and the recombination defects identified in checkpoint-deficient cells are consistent with a role of the DNA damage checkpoint in activating recombinational repair. Rad55p phosphorylation possibly affects the balance between different competing DNA repair pathways.
Figures

References
-
- Allen J B, Zhou Z, Siede W, Friedberg E C, Elledge S J. The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev. 1994;8:2401–2415. - PubMed
-
- Baumann P, West S C. Role of the human RAD51 protein in homologous recombination and double-stranded break repair. Trends Biochem Sci. 1998;23:247–251. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases