

Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I epsilon
- PMID: 10848614
- PMCID: PMC85940
- DOI: 10.1128/MCB.20.13.4888-4899.2000
Nuclear entry of the circadian regulator mPER1 is controlled by mammalian casein kinase I epsilon
Abstract
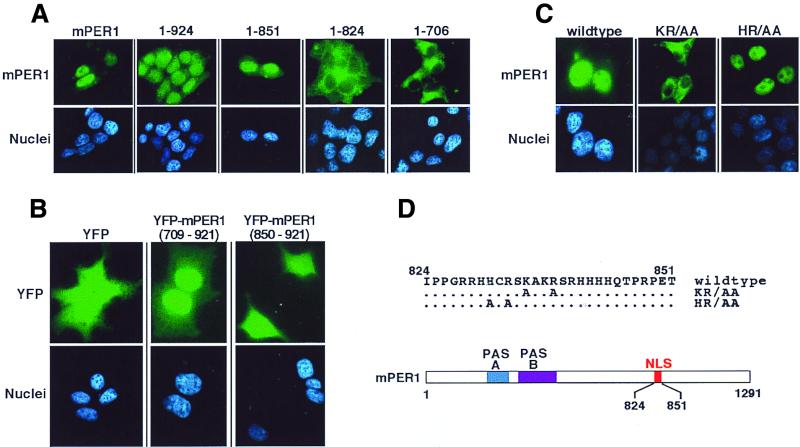
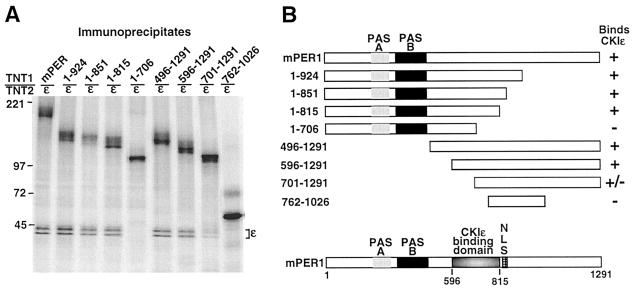
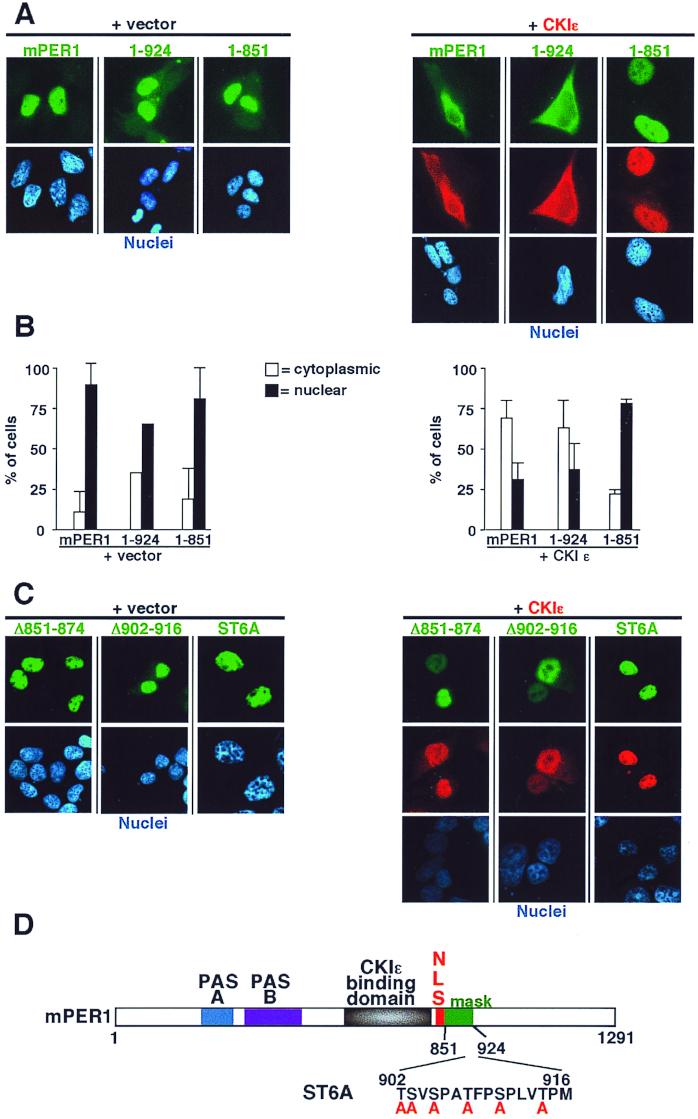
The molecular oscillator that keeps circadian time is generated by a negative feedback loop. Nuclear entry of circadian regulatory proteins that inhibit transcription from E-box-containing promoters appears to be a critical component of this loop in both Drosophila and mammals. The Drosophila double-time gene product, a casein kinase I epsilon (CKIepsilon) homolog, has been reported to interact with dPER and regulate circadian cycle length. We find that mammalian CKIepsilon binds to and phosphorylates the murine circadian regulator mPER1. Unlike both dPER and mPER2, mPER1 expressed alone in HEK 293 cells is predominantly a nuclear protein. Two distinct mechanisms appear to retard mPER1 nuclear entry. First, coexpression of mPER2 leads to mPER1-mPER2 heterodimer formation and cytoplasmic colocalization. Second, coexpression of CKIepsilon leads to masking of the mPER1 nuclear localization signal and phosphorylation-dependent cytoplasmic retention of both proteins. CKIepsilon may regulate mammalian circadian rhythm by controlling the rate at which mPER1 enters the nucleus.
Figures
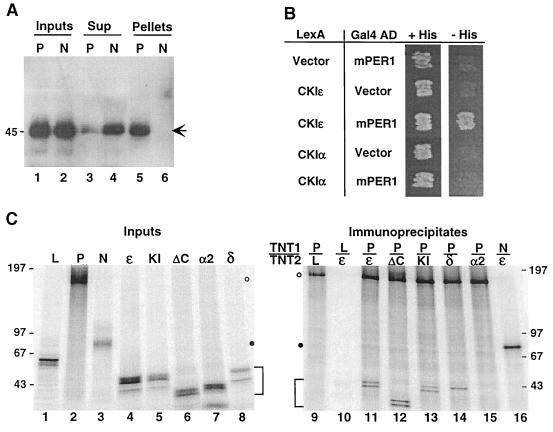
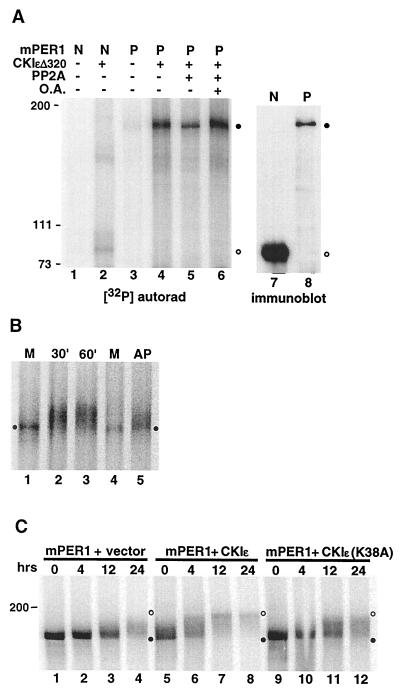
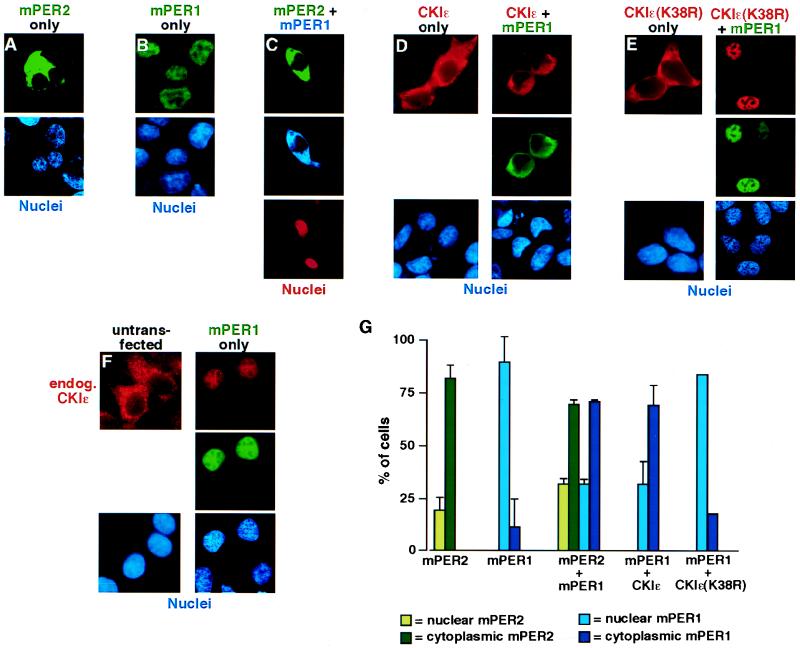
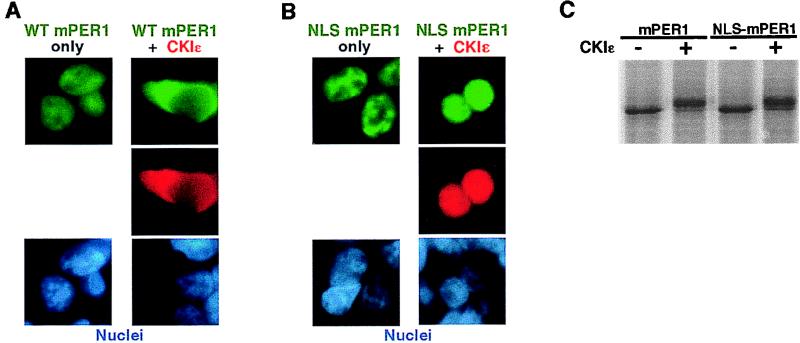
References
-
- Albrecht U, Sun Z S, Eichele G, Lee C C. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell. 1997;91:1055–1064. - PubMed
-
- Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. - PubMed
-
- Briggs L J, Stein D, Goltz J, Corrigan V C, Efthymiadis A, Hubner S, Jans D A. The cAMP-dependent protein kinase site (Ser312) enhances dorsal nuclear import through facilitating nuclear localization sequence/importin interaction. J Biol Chem. 1998;273:22745–22752. - PubMed
-
- Brunet A, Bonni A, Zigmond M J, Lin M Z, Juo P, Hu L S, Anderson M J, Arden K C, Blenis J, Greenberg M E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. - PubMed
-
- Cegielska A, Gietzen K F, Rivers A, Virshup D M. Autoinhibition of casein kinase I ɛ (CKIɛ) is relieved by protein phosphatases and limited proteolysis. J Biol Chem. 1998;273:1357. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous