

Whole-genome trees based on the occurrence of folds and orthologs: implications for comparing genomes on different levels
- PMID: 10854412
- PMCID: PMC310900
- DOI: 10.1101/gr.10.6.808
Whole-genome trees based on the occurrence of folds and orthologs: implications for comparing genomes on different levels
Abstract
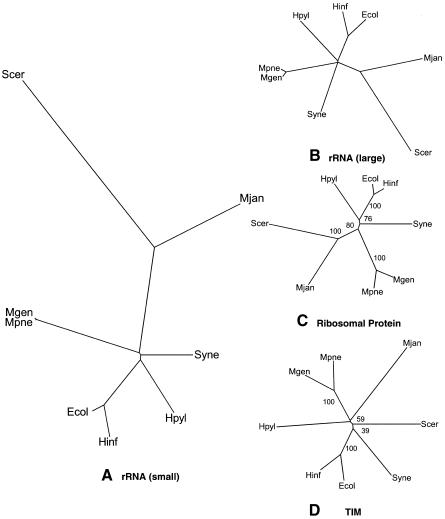
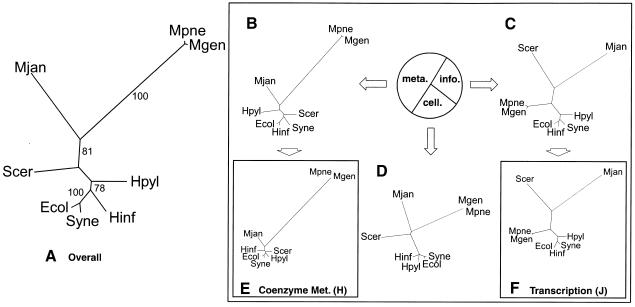
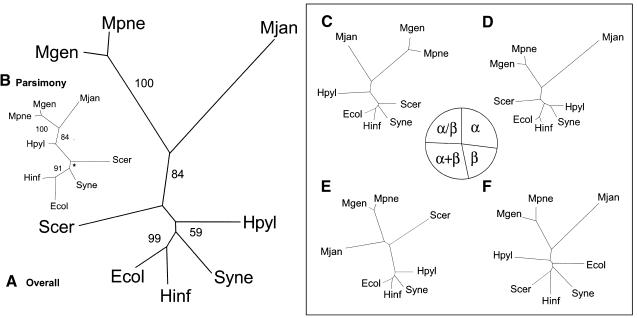
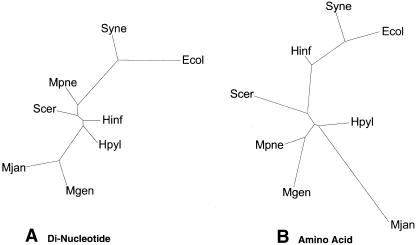
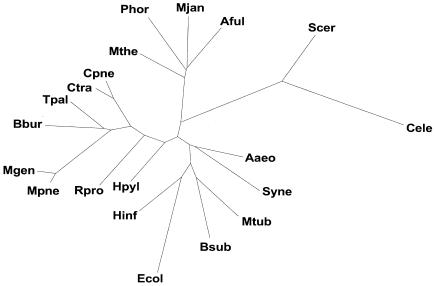
We built whole-genome trees based on the presence or absence of particular molecular features, either orthologs or folds, in the genomes of a number of recently sequenced microorganisms. To put these genomic trees into perspective, we compared them to the traditional ribosomal phylogeny and also to trees based on the sequence similarity of individual orthologous proteins. We found that our genomic trees based on the overall occurrence of orthologs did not agree well with the traditional tree. This discrepancy, however, vanished when one restricted the tree to proteins involved in transcription and translation, not including problematic proteins involved in metabolism. Protein folds unite superficially unrelated sequence families and represent a most fundamental molecular unit described by genomes. We found that our genomic occurrence tree based on folds agreed fairly well with the traditional ribosomal phylogeny. Surprisingly, despite this overall agreement, certain classes of folds, particularly all-beta ones, had a somewhat different phylogenetic distribution. We also compared our occurrence trees to whole-genome clusters based on the composition of amino acids and di-nucleotides. Finally, we analyzed some technical aspects of genomic trees-e.g., comparing parsimony versus distance-based approaches and examining the effects of increasing numbers of organisms. Additional information (e.g. clickable trees) is available from http://bioinfo.mbb.yale.edu/genome/trees.
Figures
References
-
- Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. - PubMed
-
- Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources