

Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution
- PMID: 10860959
- PMCID: PMC34372
- DOI: 10.1073/pnas.97.13.6974
Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution
Abstract
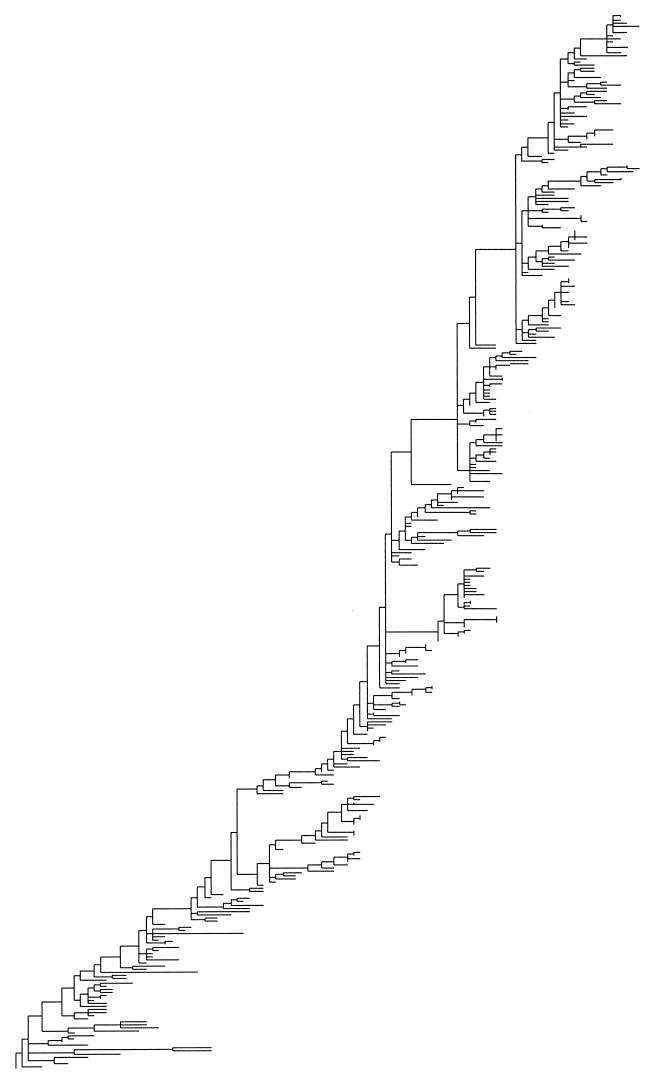
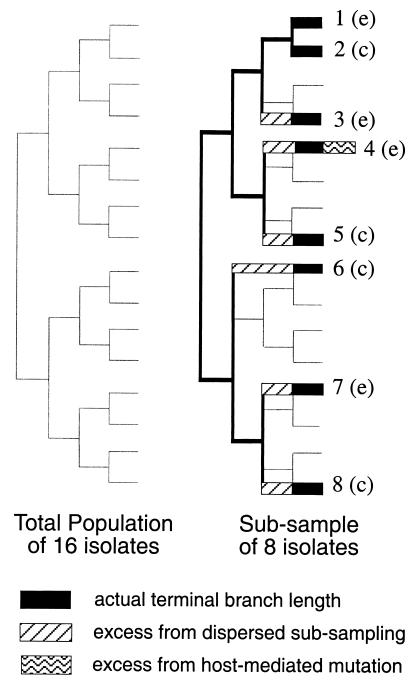
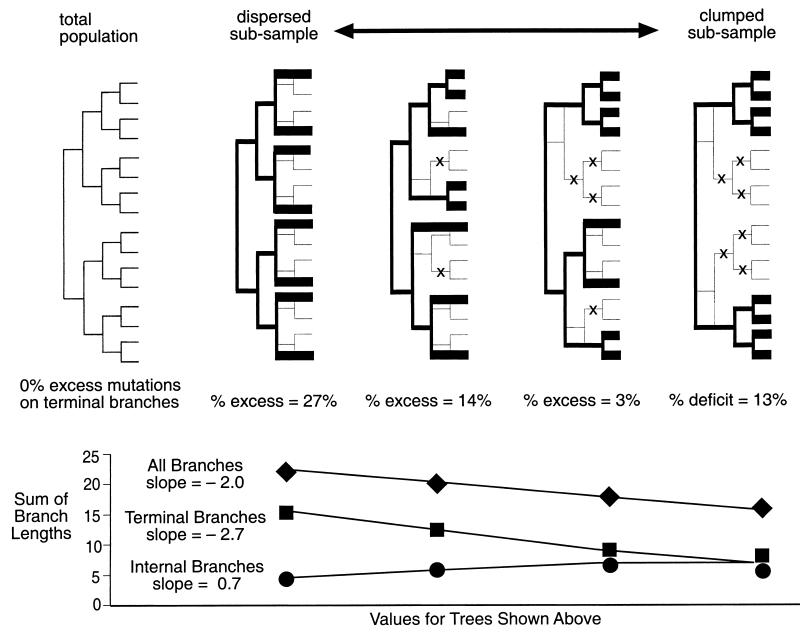
In this paper we determine the extent to which host-mediated mutations and a known sampling bias affect evolutionary studies of human influenza A. Previous phylogenetic reconstruction of influenza A (H3N2) evolution using the hemagglutinin gene revealed an excess of nonsilent substitutions assigned to the terminal branches of the tree. We investigate two hypotheses to explain this observation. The first hypothesis is that the excess reflects mutations that were either not present or were at low frequency in the viral sample isolated from its human host, and that these mutations increased in frequency during passage of the virus in embryonated eggs. A set of 22 codons known to undergo such "host-mediated" mutations showed a significant excess of mutations assigned to branches attaching sequences from egg-cultured (as opposed to cell-cultured) isolates to the tree. Our second hypothesis is that the remaining excess results from sampling bias. Influenza surveillance is purposefully biased toward sequencing antigenically dissimilar strains in an effort to identify new variants that may signal the need to update the vaccine. This bias produces an excess of mutations assigned to terminal branches simply because an isolate with no close relatives is by definition attached to the tree by a relatively long branch. Simulations show that the magnitude of excess mutations we observed in the hemagglutinin tree is consistent with expectations based on our sampling protocol. Sampling bias does not affect inferences about evolution drawn from phylogenetic analyses. However, if possible, the excess caused by host-mediated mutations should be removed from studies of the evolution of influenza viruses as they replicate in their human hosts.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
