

Sequences of Citrus tristeza virus separated in time and space are essentially identical
- PMID: 10888625
- PMCID: PMC112203
- DOI: 10.1128/jvi.74.15.6856-6865.2000
Sequences of Citrus tristeza virus separated in time and space are essentially identical
Abstract
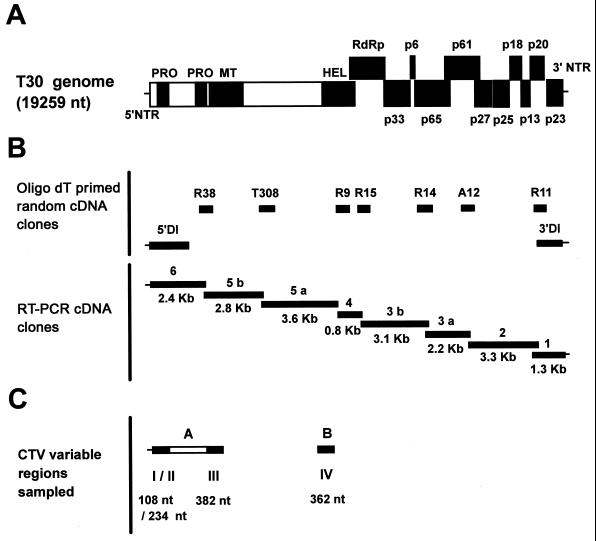
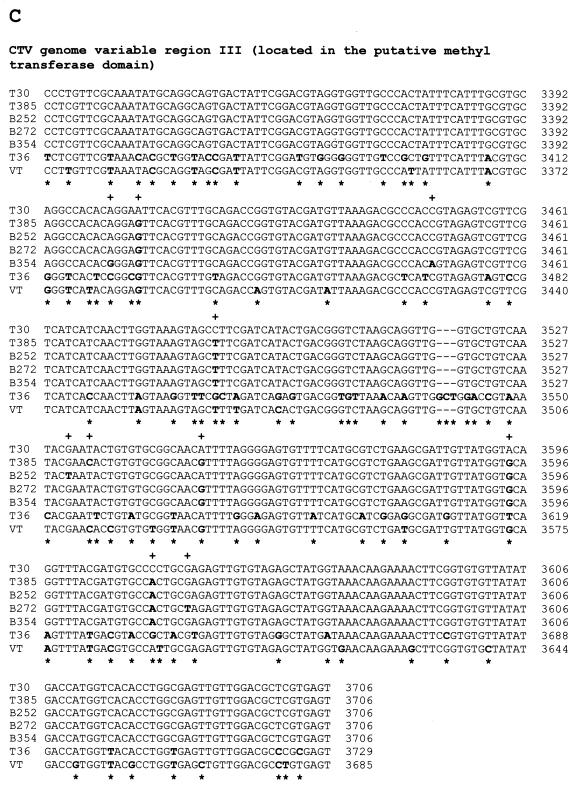
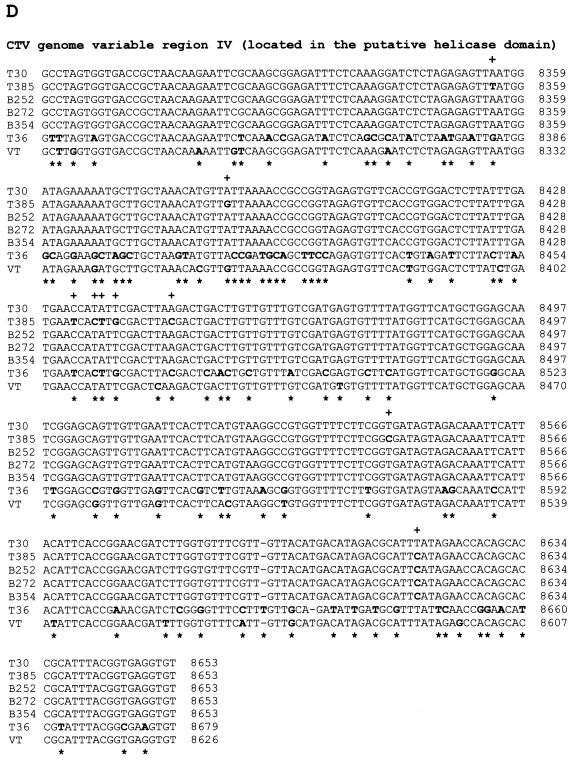
The first Citrus tristeza virus (CTV) genomes completely sequenced (19.3-kb positive-sense RNA), from four biologically distinct isolates, are unexpectedly divergent in nucleotide sequence (up to 60% divergence). Understanding of whether these large sequence differences resulted from recent evolution is important for the design of disease management strategies, particularly the use of genetically engineered mild (essentially symptomless)-strain cross protection and RNA-mediated transgenic resistance. The complete sequence of a mild isolate (T30) which has been endemic in Florida for about a century was found to be nearly identical to the genomic sequence of a mild isolate (T385) from Spain. Moreover, samples of sequences of other isolates from distinct geographic locations, maintained in different citrus hosts and also separated in time (B252 from Taiwan, B272 from Colombia, and B354 from California), were nearly identical to the T30 sequence. The sequence differences between these isolates were within or near the range of variability of the T30 population. A possible explanation for these results is that the parents of isolates T30, T385, B252, B272, and B354 have a common origin, probably Asia, and have changed little since they were dispersed throughout the world by the movement of citrus. Considering that the nucleotide divergence among the other known CTV genomes is much greater than those expected for strains of the same virus, the remarkable similarity of these five isolates indicates a high degree of evolutionary stasis in some CTV populations.
Figures


References
-
- Albiach-Martí R M, Guerri J, Hermoso de Mendoza A, Laigret F, Ballester Olmos J F, Moreno P. Aphid transmission alters the genomic and defective RNA populations of citrus tristeza virus isolates. Phytopathology. 1999;90:134–138. - PubMed
-
- Ayllón M A, Rubio L, Moya A, Guerri J, Moreno P. The haplotype distribution of two genes of citrus tristeza virus is altered after host change or aphid transmission. Virology. 1999;255:32–39. - PubMed
-
- Bar-Joseph M, Marcus R, Lee R F. The continuous challenge of citrus tristeza virus control. Annu Rev Phytopathol. 1989;27:291–316.
-
- Blok J, Mackenzie A, Guy P, Gibbs A. Nucleotide sequence comparisons of turnip yellow mosaic virus from Australia and Europe. Arch Virol. 1987;97:283–295. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
