

Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding
- PMID: 10890912
- PMCID: PMC26930
- DOI: 10.1073/pnas.150220297
Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding
Abstract
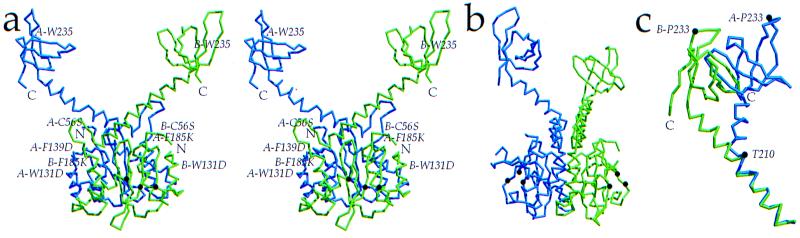
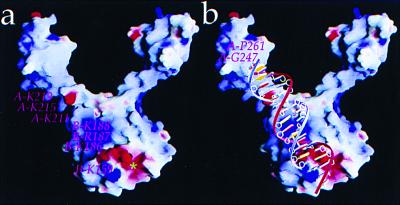
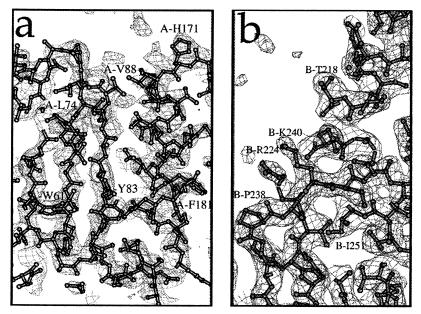
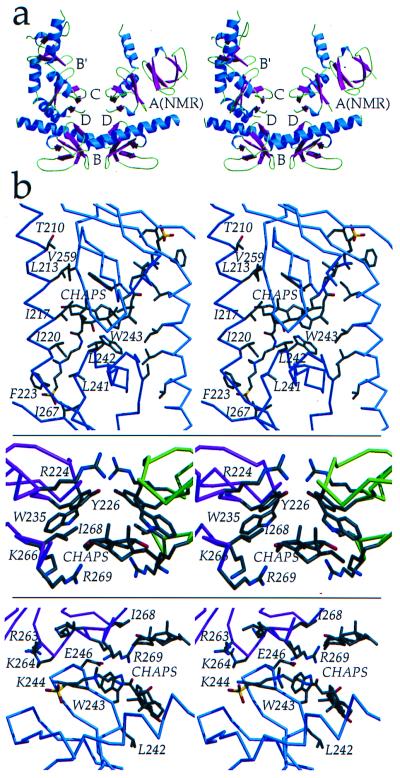
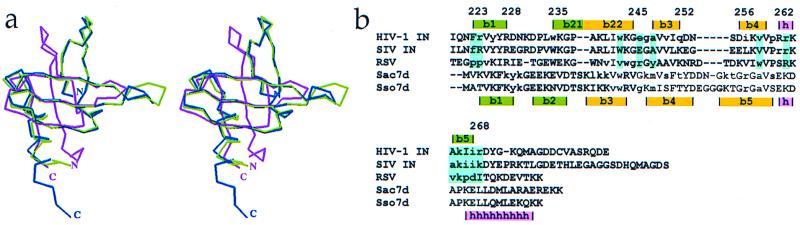
Insolubility of full-length HIV-1 integrase (IN) limited previous structure analyses to individual domains. By introducing five point mutations, we engineered a more soluble IN that allowed us to generate multidomain HIV-1 IN crystals. The first multidomain HIV-1 IN structure is reported. It incorporates the catalytic core and C-terminal domains (residues 52-288). The structure resolved to 2.8 A is a Y-shaped dimer. Within the dimer, the catalytic core domains form the only dimer interface, and the C-terminal domains are located 55 A apart. A 26-aa alpha-helix, alpha6, links the C-terminal domain to the catalytic core. A kink in one of the two alpha6 helices occurs near a known proteolytic site, suggesting that it may act as a flexible elbow to reorient the domains during the integration process. Two proteins that bind DNA in a sequence-independent manner are structurally homologous to the HIV-1 IN C-terminal domain, suggesting a similar protein-DNA interaction in which the IN C-terminal domain may serve to bind, bend, and orient viral DNA during integration. A strip of positively charged amino acids contributed by both monomers emerges from each active site of the dimer, suggesting a minimally dimeric platform for binding each viral DNA end. The crystal structure of the isolated catalytic core domain (residues 52-210), independently determined at 1.6-A resolution, is identical to the core domain within the two-domain 52-288 structure.
Figures
References
-
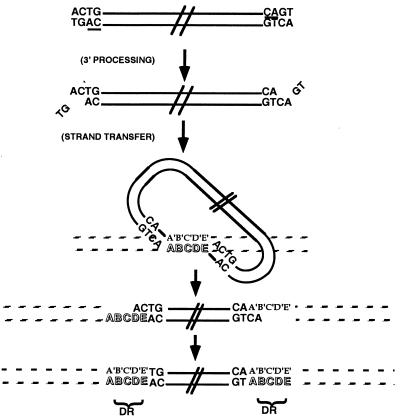
- Engelman A, Mizuuchi K, Craigie R. Cell. 1991;67:1211–1221. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
