

Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells
- PMID: 10900013
- PMCID: PMC26984
- DOI: 10.1073/pnas.97.15.8542
Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells
Abstract
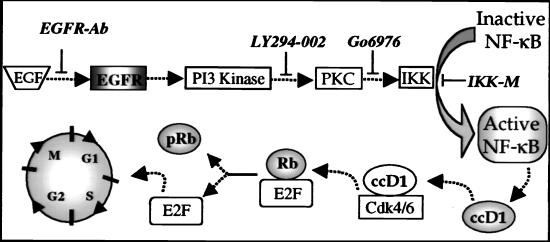
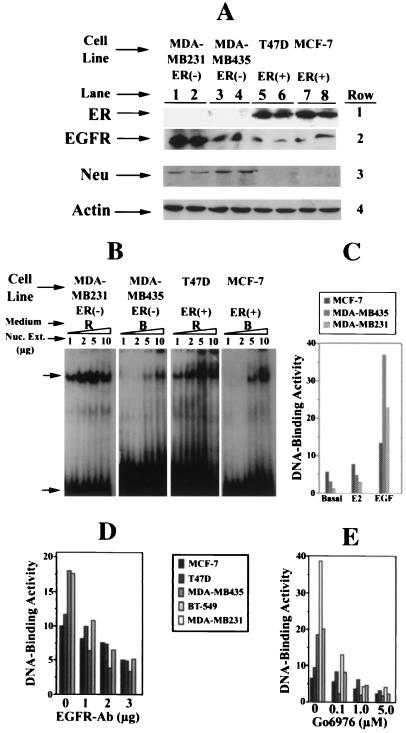
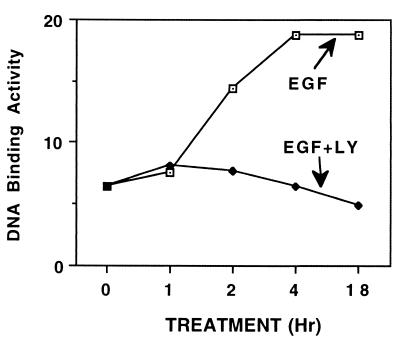
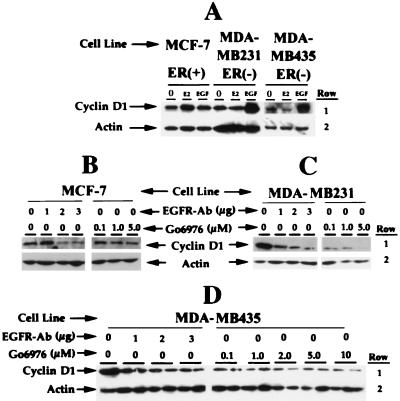
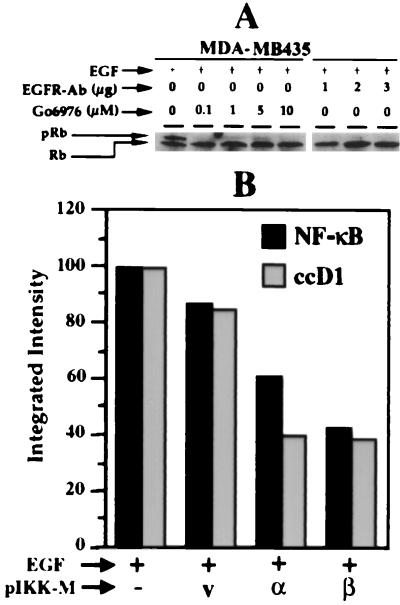
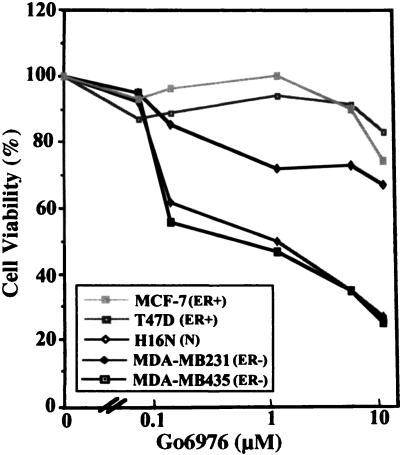
The epidermal growth factor (EGF) family of receptors (EGFR) is overproduced in estrogen receptor (ER) negative (-) breast cancer cells. An inverse correlation of the level of EGFR and ER is observed between ER- and ER positive (+) breast cancer cells. A comparative study with EGFR-overproducing ER- and low-level producing ER+ breast cancer cells suggests that EGF is a major growth-stimulating factor for ER- cells. An outline of the pathway for the EGF-induced enhanced proliferation of ER- human breast cancer cells is proposed. The transmission of mitogenic signal induced by EGF-EGFR interaction is mediated via activation of nuclear factor kappaB (NF-kappaB). The basal level of active NF-kappaB in ER- cells is elevated by EGF and inhibited by anti-EGFR antibody (EGFR-Ab), thus qualifying EGF as a NF-kappaB activation factor. NF-kappaB transactivates the cell-cycle regulatory protein, cyclin D1, which causes increased phosphorylation of retinoblastoma protein, more strongly in ER- cells. An inhibitor of phosphatidylinositol 3 kinase, Ly294-002, blocked this event, suggesting a role of the former in the activation of NF-kappaB by EGF. Go6976, a well-characterized NF-kappaB inhibitor, blocked EGF-induced NF-kappaB activation and up-regulation of cell-cycle regulatory proteins. This low molecular weight compound also caused apoptotic death, predominantly more in ER- cells. Thus Go6976 and similar NF-kappaB inhibitors are potentially novel low molecular weight therapeutic agents for treatment of ER- breast cancer patients.
Figures
References
-
- Hortobagyi G N. N Engl J Med. 1998;339:974–984. - PubMed
-
- Brown M. Hematol Oncol Clin North Am. 1994;8:101–112. - PubMed
-
- Jensen E V. Breast Cancer Res Treat. 1981;26:2319–2326. - PubMed
-
- Jordan V C. Breast Cancer Res Treat. 1995;36:267–285. - PubMed
-
- Smith D F, Toft D O. Mol Endocrinol. 1993;7:4–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
