

Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells
- PMID: 10906200
- PMCID: PMC112267
- DOI: 10.1128/jvi.74.16.7470-7477.2000
Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells
Abstract
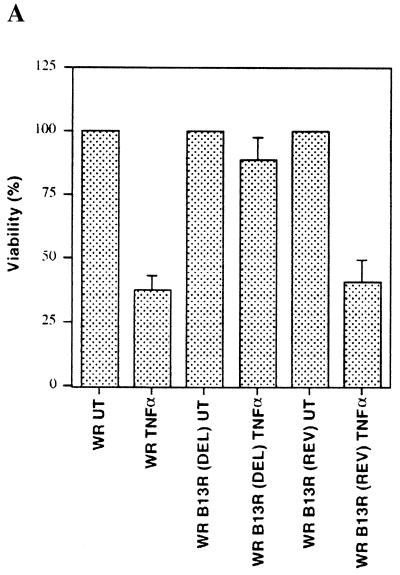
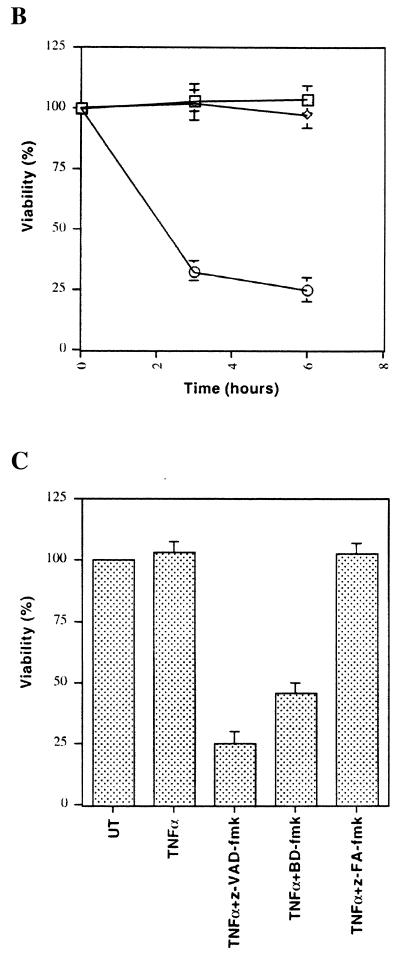
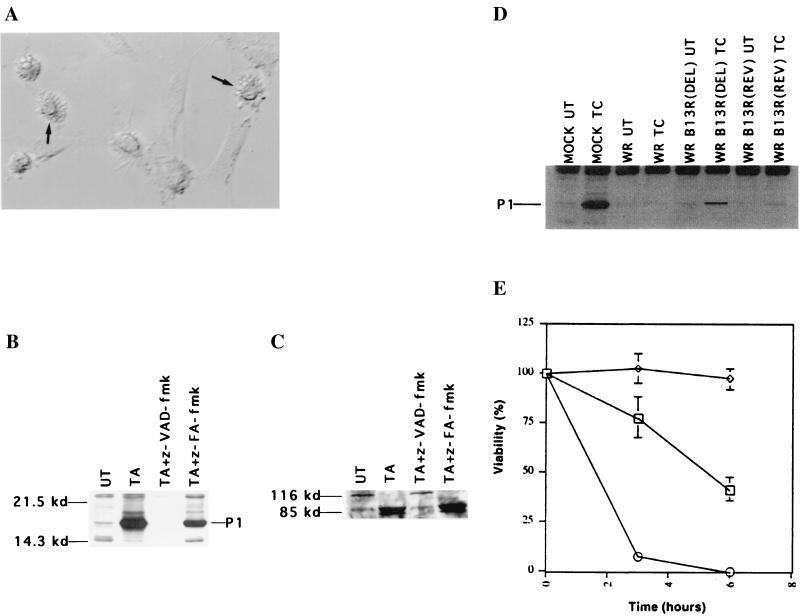
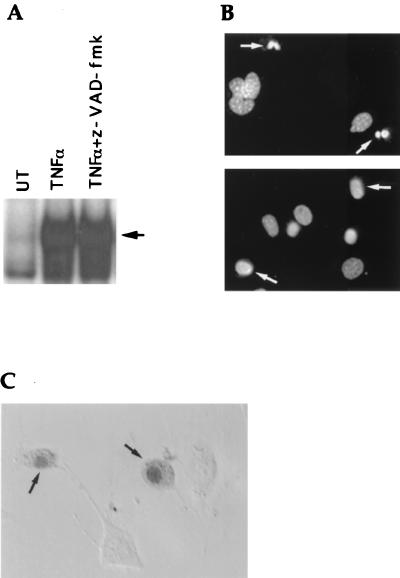
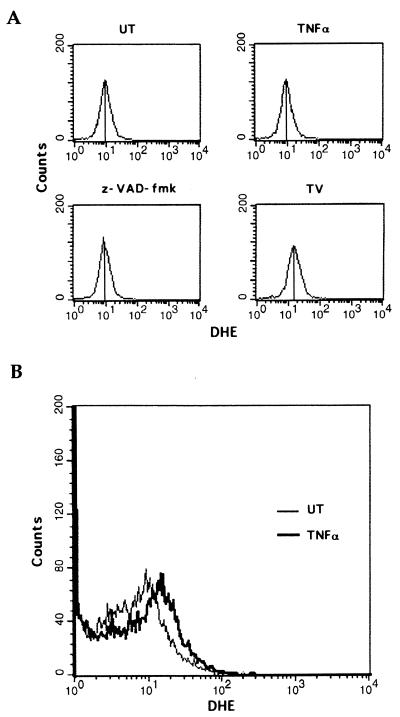
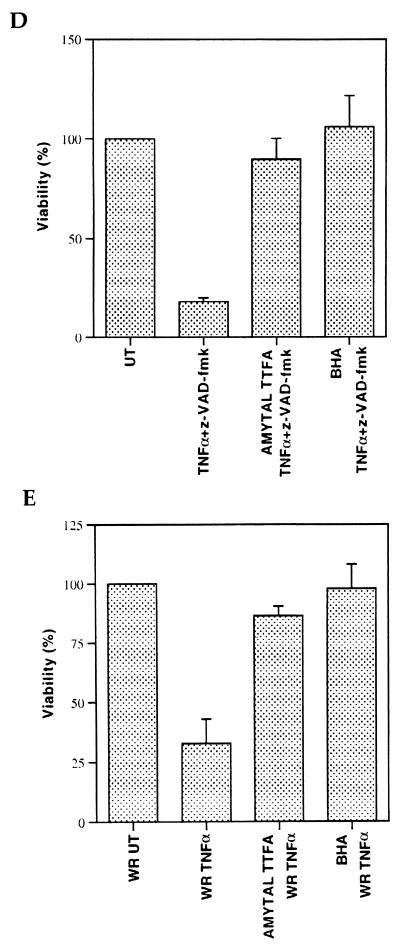
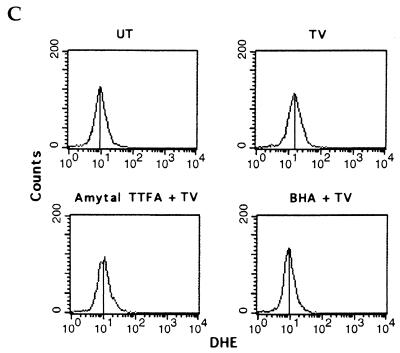
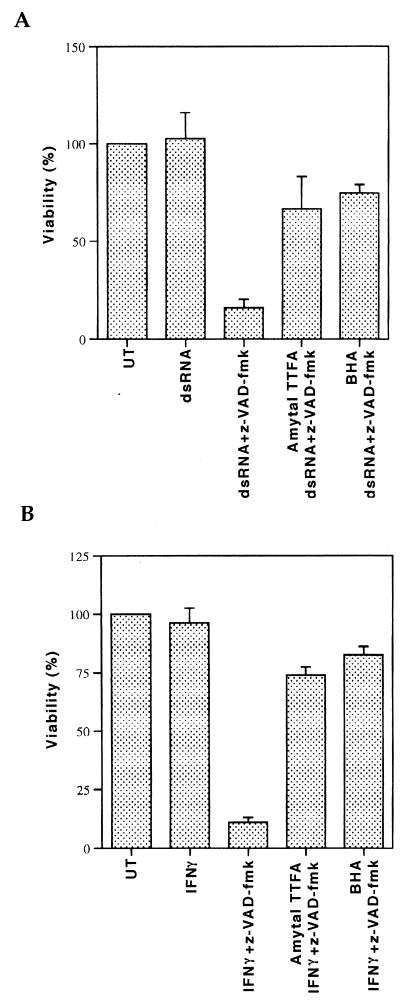
Induction of apoptotic cell death generally requires the participation of cysteine proteases belonging to the caspase family. However, and similar to most cell types, mouse fibroblasts are normally resistant to tumor necrosis factor alpha (TNF-alpha)-induced apoptosis. Surprisingly, TNF-alpha treatment of vaccinia virus-infected mouse fibroblasts resulted in necrotic-like cell death, which was significantly reduced in cells infected with a vaccinia virus mutant lacking the caspase inhibitor B13R. Furthermore, TNF-alpha also induced necrotic-like cell death of fibroblasts in the presence of peptidyl caspase inhibitors. In both cases, necrosis was accompanied by generation of superoxide species. Caspase inhibitors also sensitized fibroblasts to killing by double-stranded RNA and gamma interferon. In all cases, cell death was efficiently blocked by antioxidants or mitochondrial respiratory chain inhibitors. These results define a new mitochondrion-dependent mechanism which may be important in the killing of cells infected with viruses encoding caspase inhibitors.
Figures
References
-
- Ashkenazi A, Dixit V M. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. - PubMed
-
- Beg A A, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. - PubMed
-
- Beg A A, Sha W C, Bronson R T, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. - PubMed
-
- Boehm U, Klamp T, Groot M, Howard J C. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
