

The endonuclease activity of the yeast Dna2 enzyme is essential in vivo
- PMID: 10908349
- PMCID: PMC102684
- DOI: 10.1093/nar/28.15.2873
The endonuclease activity of the yeast Dna2 enzyme is essential in vivo
Abstract
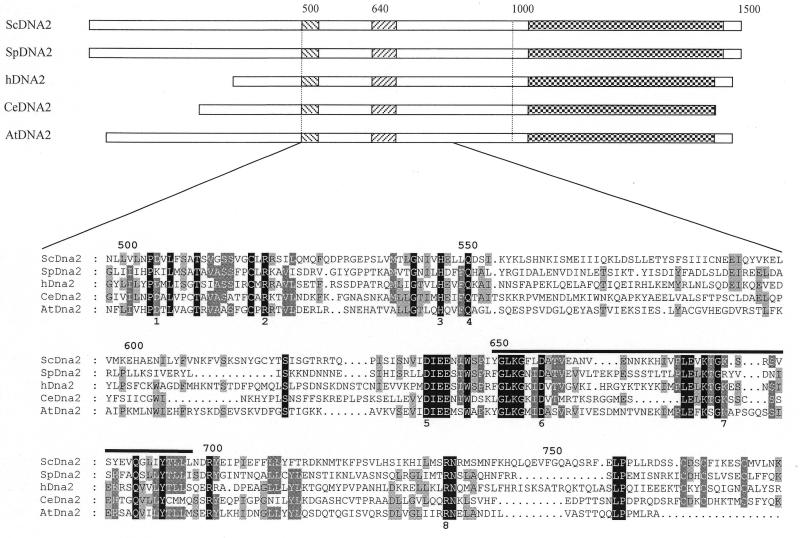
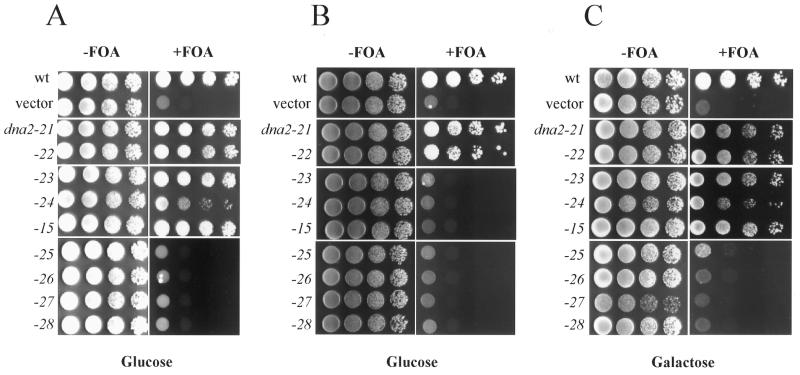
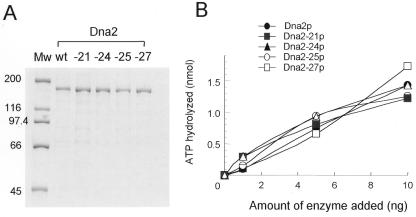
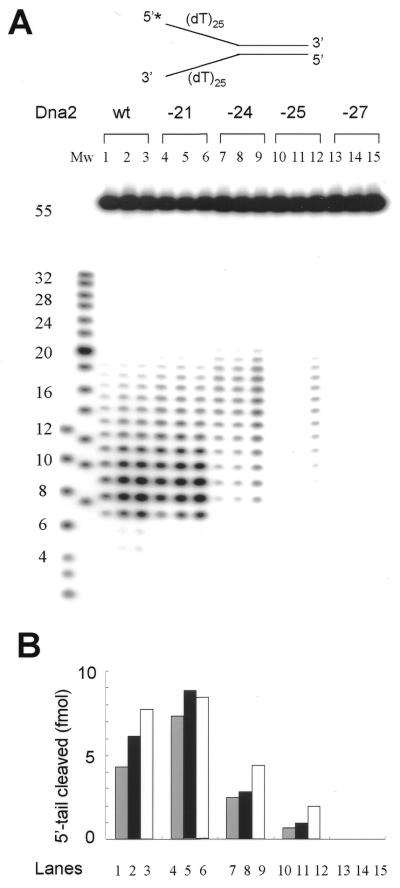
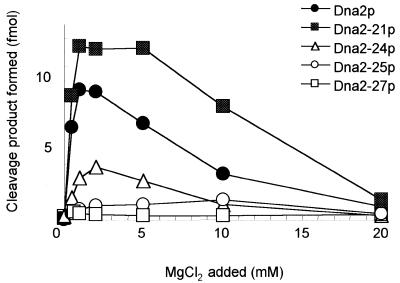
Dna2 is a multifunctional enzyme in yeast that possesses endonuclease activity well suited to remove RNA-DNA primers of Okazaki fragments, raising the question of whether endonuclease activity is essential for in vivo Dna2 function. Systematic site-directed mutations of amino acid residues in Saccharomyces cerevisiae DNA2 conserved in the central region of many eukaryotic DNA2 homologs allowed us to identify mutant dna2 alleles that were divided into three groups based on the viability of the mutant cells: (i) viable; (ii) inviable only when expression was repressed; (iii) inviable. Biochemical analyses of recombinant mutant Dna2 proteins isolated from the latter two groups revealed that they possessed normal ATPase/helicase activity, but were impaired in their endonuclease activity. Cells expressing mutant Dna2 enzymes partially impaired in endonuclease activity were viable, but were unable to grow when expression of their mutant Dna2 enzymes was further reduced. Their growth was restored when the mutant Dna2 proteins decreased in nuclease activity were induced to overexpress. In contrast, mutant Dna2 proteins lacking endonuclease activity did not allow cells to grow under any conditions tested. These in vivo and in vitro results demonstrate that the endonuclease activity of Dna2 is essential for Okazaki fragment processing.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
