

Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8
- PMID: 10921868
- PMCID: PMC306600
- DOI: 10.1093/emboj/19.15.3857
Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8
Abstract
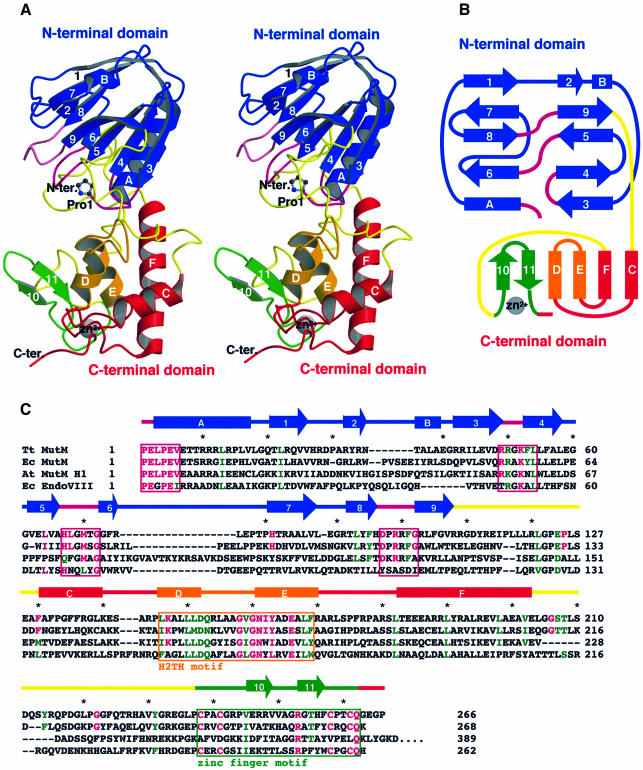
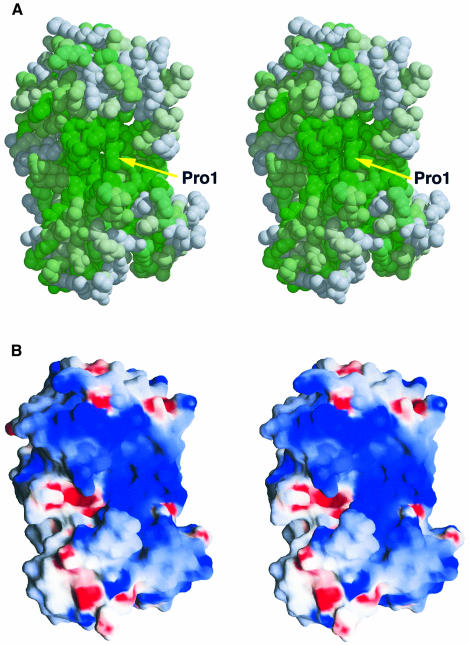
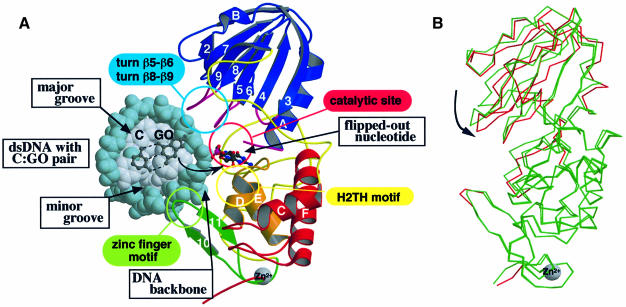
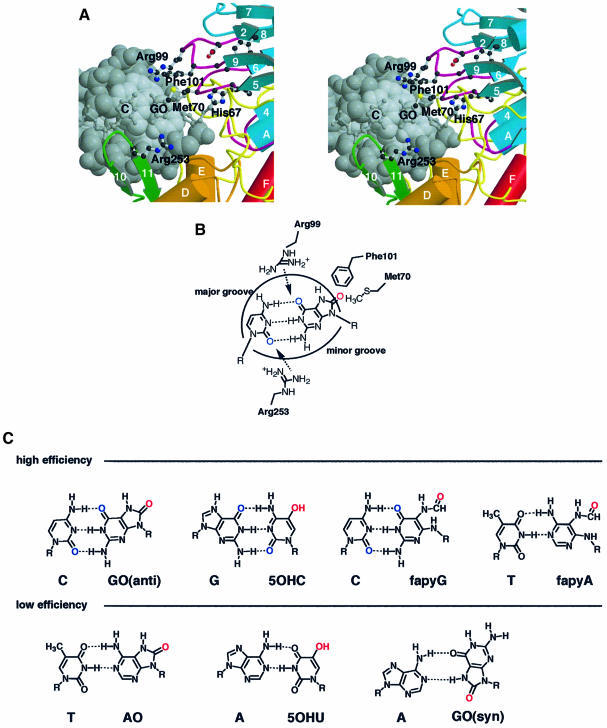
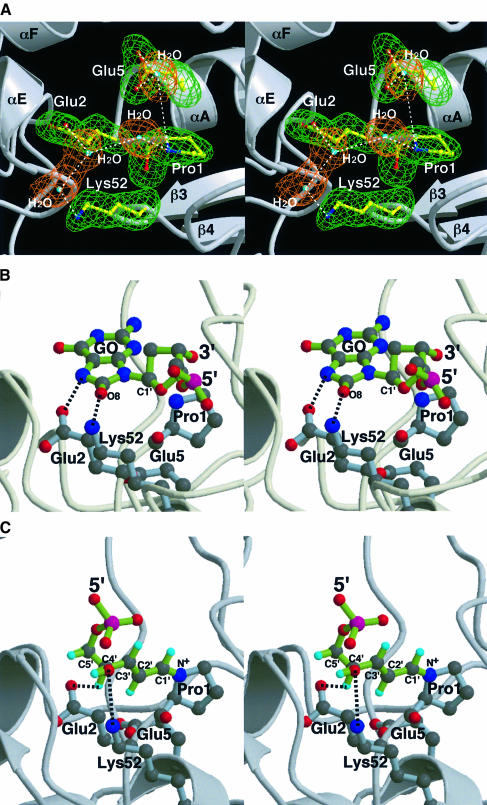
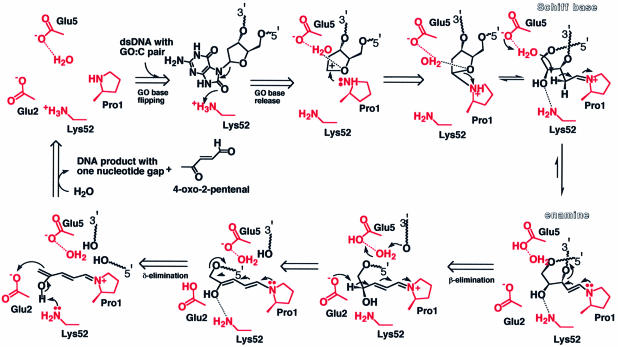
The MutM [formamidopyrimidine DNA glycosylase (Fpg)] protein is a trifunctional DNA base excision repair enzyme that removes a wide range of oxidatively damaged bases (N-glycosylase activity) and cleaves both the 3'- and 5'-phosphodiester bonds of the resulting apurinic/apyrimidinic site (AP lyase activity). The crystal structure of MutM from an extreme thermophile, Thermus thermophilus HB8, was determined at 1.9 A resolution with multiwavelength anomalous diffraction phasing using the intrinsic Zn(2+) ion of the zinc finger. MutM is composed of two distinct and novel domains connected by a flexible hinge. There is a large, electrostatically positive cleft lined by highly conserved residues between the domains. On the basis of the three-dimensional structure and taking account of previous biochemical experiments, we propose a DNA-binding mode and reaction mechanism for MutM. The locations of the putative catalytic residues and the two DNA-binding motifs (the zinc finger and the helix-two-turns-helix motifs) suggest that the oxidized base is flipped out from double-stranded DNA in the binding mode and excised by a catalytic mechanism similar to that of bifunctional base excision repair enzymes.
Figures

Similar articles
-
Thermostable repair enzyme for oxidative DNA damage from extremely thermophilic bacterium, Thermus thermophilus HB8.Nucleic Acids Res. 1998 Feb 15;26(4):903-10. doi: 10.1093/nar/26.4.903. Nucleic Acids Res. 1998. PMID: 9461446 Free PMC article.
-
Structural insights into lesion recognition and repair by the bacterial 8-oxoguanine DNA glycosylase MutM.Nat Struct Biol. 2002 Jul;9(7):544-52. doi: 10.1038/nsb809. Nat Struct Biol. 2002. PMID: 12055620
-
Crystallization and preliminary X-ray crystallographic studies of Thermus thermophilus HB8 MutM protein involved in repairs of oxidative DNA damage.J Biochem. 2000 Jan;127(1):9-11. doi: 10.1093/oxfordjournals.jbchem.a022588. J Biochem. 2000. PMID: 10731660
-
Repair of oxidative DNA damage: mechanisms and functions.Cell Biochem Biophys. 2001;35(2):141-70. doi: 10.1385/CBB:35:2:141. Cell Biochem Biophys. 2001. PMID: 11892789 Review.
-
The OGG1 gene encodes a repair enzyme for oxidatively damaged DNA and is involved in human carcinogenesis.Antioxid Redox Signal. 2001 Aug;3(4):597-609. doi: 10.1089/15230860152542952. Antioxid Redox Signal. 2001. PMID: 11554447 Review.
Cited by
-
Structural Aspects of DNA Repair and Recombination in Crop Improvement.Front Genet. 2020 Sep 11;11:574549. doi: 10.3389/fgene.2020.574549. eCollection 2020. Front Genet. 2020. PMID: 33024442 Free PMC article. Review.
-
Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA.Proc Natl Acad Sci U S A. 2002 Mar 19;99(6):3523-8. doi: 10.1073/pnas.062053799. Proc Natl Acad Sci U S A. 2002. PMID: 11904416 Free PMC article.
-
Recognition of a tandem lesion by DNA bacterial formamidopyrimidine glycosylases explored combining molecular dynamics and machine learning.Comput Struct Biotechnol J. 2021 Apr 30;19:2861-2869. doi: 10.1016/j.csbj.2021.04.055. eCollection 2021. Comput Struct Biotechnol J. 2021. PMID: 34093997 Free PMC article.
-
A novel nucleoid-associated protein specific to the actinobacteria.Nucleic Acids Res. 2013 Apr;41(7):4171-84. doi: 10.1093/nar/gkt095. Epub 2013 Feb 20. Nucleic Acids Res. 2013. PMID: 23427309 Free PMC article.
-
The Fpg/Nei family of DNA glycosylases: substrates, structures, and search for damage.Prog Mol Biol Transl Sci. 2012;110:71-91. doi: 10.1016/B978-0-12-387665-2.00004-3. Prog Mol Biol Transl Sci. 2012. PMID: 22749143 Free PMC article. Review.
References
-
- Bhagwat M. and Gerlt,J.A. (1996) 3′- and 5′-strand cleavage reactions catalyzed by the Fpg protein from Escherichia coli occur via successive β- and δ-elimination mechanisms, respectively. Biochemistry, 35, 659–665. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
