

Identification and analysis of error types in high-throughput genotyping
- PMID: 10924406
- PMCID: PMC1287531
- DOI: 10.1086/303048
Identification and analysis of error types in high-throughput genotyping
Abstract
Although it is clear that errors in genotyping data can lead to severe errors in linkage analysis, there is as yet no consensus strategy for identification of genotyping errors. Strategies include comparison of duplicate samples, independent calling of alleles, and Mendelian-inheritance-error checking. This study aimed to develop a better understanding of error types associated with microsatellite genotyping, as a first step toward development of a rational error-detection strategy. Two microsatellite marker sets (a commercial genomewide set and a custom-designed fine-resolution mapping set) were used to generate 118,420 and 22,500 initial genotypes and 10,088 and 8,328 duplicates, respectively. Mendelian-inheritance errors were identified by PedManager software, and concordance was determined for the duplicate samples. Concordance checking identifies only human errors, whereas Mendelian-inheritance-error checking is capable of detection of additional errors, such as mutations and null alleles. Neither strategy is able to detect all errors. Inheritance checking of the commercial marker data identified that the results contained 0.13% human errors and 0.12% other errors (0.25% total error), whereas concordance checking found 0.16% human errors. Similarly, Mendelian-inheritance-error checking of the custom-set data identified 1.37% errors, compared with 2.38% human errors identified by concordance checking. A greater variety of error types were detected by Mendelian-inheritance-error checking than by duplication of samples or by independent reanalysis of gels. These data suggest that Mendelian-inheritance-error checking is a worthwhile strategy for both types of genotyping data, whereas fine-mapping studies benefit more from concordance checking than do studies using commercial marker data. Maximization of error identification increases the likelihood of linkage when complex diseases are analyzed.
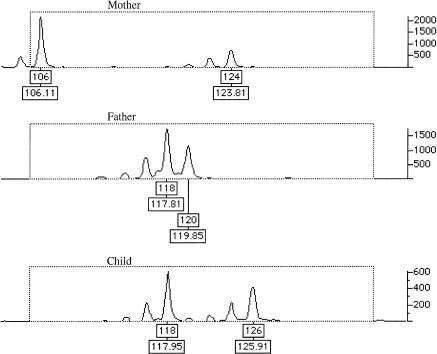
Figures


References
Electronic-Database Information
-
- Australian Genome Research Facility, http://www.agrf.org.au/ (for PedManager version 0.9)
-
- Division of Statistical Genetics, Department of Human Genetics, University of Pittsburgh, http://watson.hgen.pitt.edu/register/soft_doc.html (for PedCheck)
References
-
- Bahlo M, Broman KW (1999) Identification of and adjustment for genotyping errors in data on sibpairs when parental genotypes are unavailable. Am J Hum Genet Suppl 65:A241
-
- Bowcock AM, Ruix-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL (1994) High resolution of human evolutionary trees with polymorphic microsatellites. Nature 368:455–457 - PubMed
-
- Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20:1004–1010 - PubMed
-
- Dixon MJ, Dixon J, Raskova D, Le Beau MM, Williamson R, Klinger C, Landes GM (1992) Genetic and physical mapping of the Treacher Collins syndrome locus: refinement of the localisation to chromosome 5q 32-33.2.1. Hum Mol Genet 1:249–253 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
