

Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia
- PMID: 10930361
- PMCID: PMC1287515
- DOI: 10.1086/303056
Iron-dependent self-assembly of recombinant yeast frataxin: implications for Friedreich ataxia
Abstract
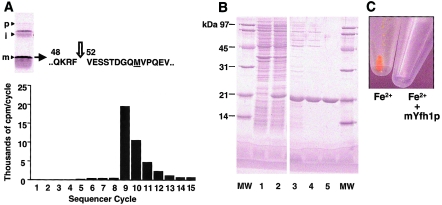
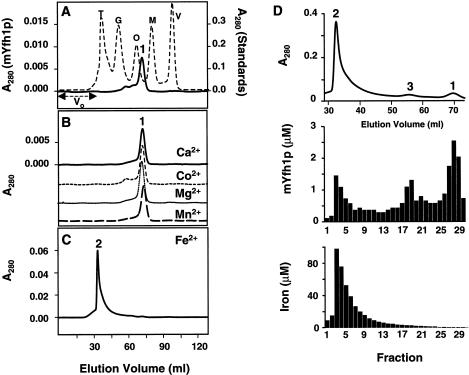
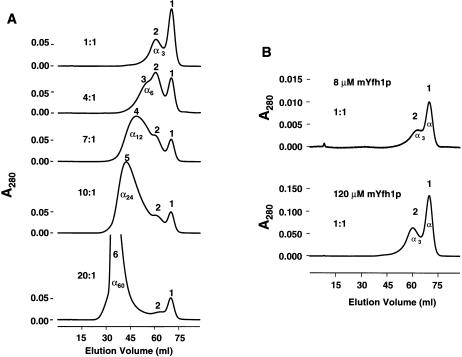
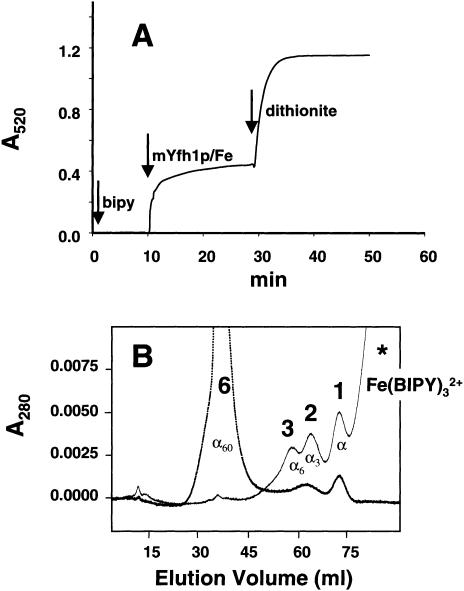
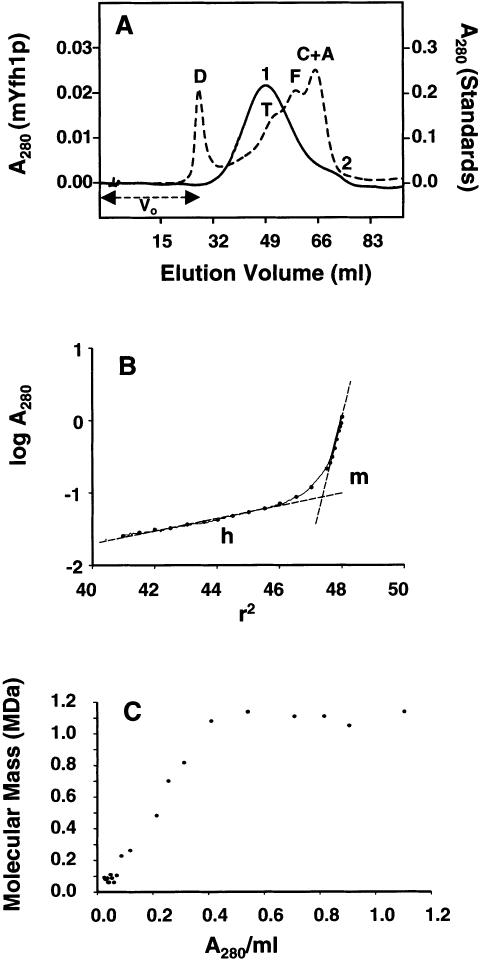
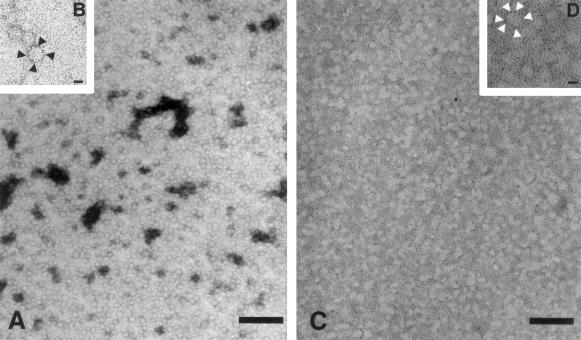
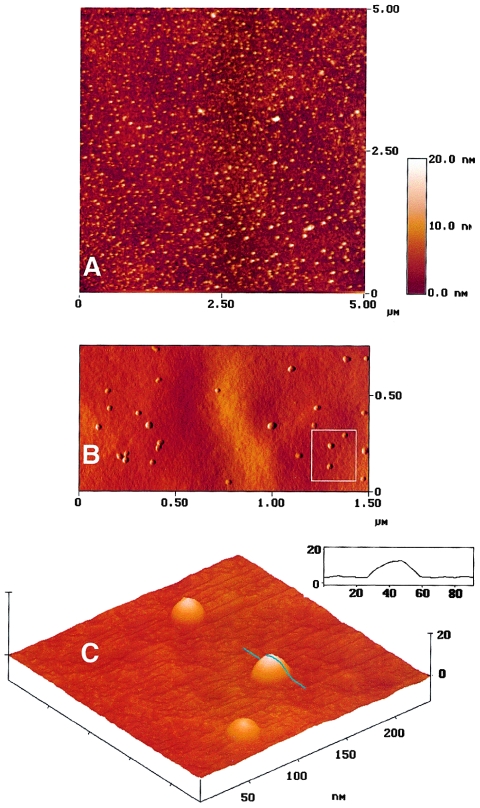
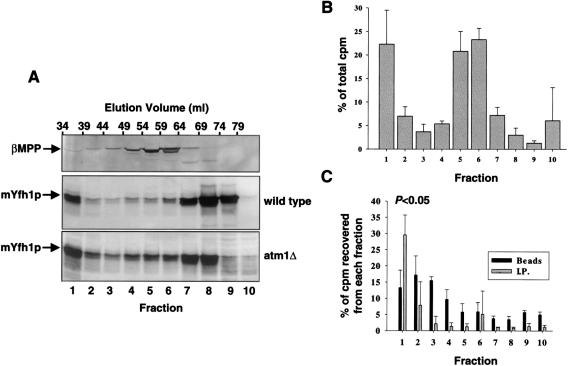
Frataxin deficiency is the primary cause of Friedreich ataxia (FRDA), an autosomal recessive cardiodegenerative and neurodegenerative disease. Frataxin is a nuclear-encoded mitochondrial protein that is widely conserved among eukaryotes. Genetic inactivation of the yeast frataxin homologue (Yfh1p) results in mitochondrial iron accumulation and hypersensitivity to oxidative stress. Increased iron deposition and evidence of oxidative damage have also been observed in cardiac tissue and cultured fibroblasts from patients with FRDA. These findings indicate that frataxin is essential for mitochondrial iron homeostasis and protection from iron-induced formation of free radicals. The functional mechanism of frataxin, however, is still unknown. We have expressed the mature form of Yfh1p (mYfh1p) in Escherichia coli and have analyzed its function in vitro. Isolated mYfh1p is a soluble monomer (13,783 Da) that contains no iron and shows no significant tendency to self-associate. Aerobic addition of ferrous iron to mYfh1p results in assembly of regular spherical multimers with a molecular mass of approximately 1. 1 MDa (megadaltons) and a diameter of 13+/-2 nm. Each multimer consists of approximately 60 subunits and can sequester >3,000 atoms of iron. Titration of mYfh1p with increasing iron concentrations supports a stepwise mechanism of multimer assembly. Sequential addition of an iron chelator and a reducing agent results in quantitative iron release with concomitant disassembly of the multimer, indicating that mYfh1p sequesters iron in an available form. In yeast mitochondria, native mYfh1p exists as monomer and a higher-order species with a molecular weight >600,000. After addition of (55)Fe to the medium, immunoprecipitates of this species contain >16 atoms of (55)Fe per molecule of mYfh1p. We propose that iron-dependent self-assembly of recombinant mYfh1p reflects a physiological role for frataxin in mitochondrial iron sequestration and bioavailability.
Figures
References
Electronic-Database Information
-
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/index.html (for YFH1 [accession number Z74168])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for FRDA [MIM 229300]) - PubMed
References
-
- Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homologue of frataxin. Science 276:1709–1712 - PubMed
-
- Bradley JL, Blake JC, Chamberlain S, Thomas PK, Cooper JM, Schapira AH (2000) Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum Mol Genet 9:275–282 - PubMed
-
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G (1999) Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J Biol Chem 274:22763–22769 - PubMed
-
- Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Durr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M (1997) Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet 6:1771–1780 - PubMed
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, et al (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271:1423–1427 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
