

An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice
- PMID: 10944233
- PMCID: PMC16936
- DOI: 10.1073/pnas.97.17.9747
An endothelium-derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium-dependent vasodilator in resistance vessels of wild-type and endothelial NO synthase knockout mice
Abstract
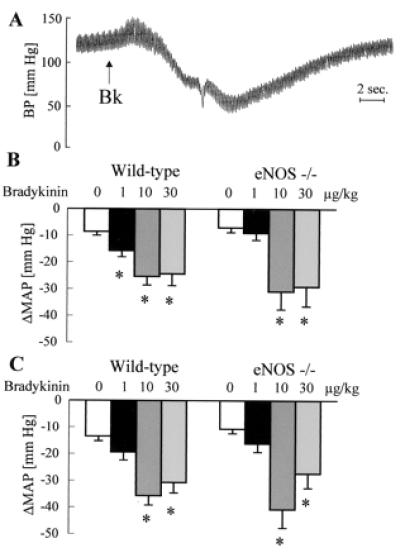
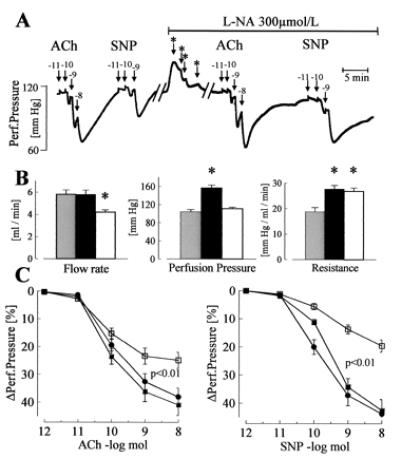
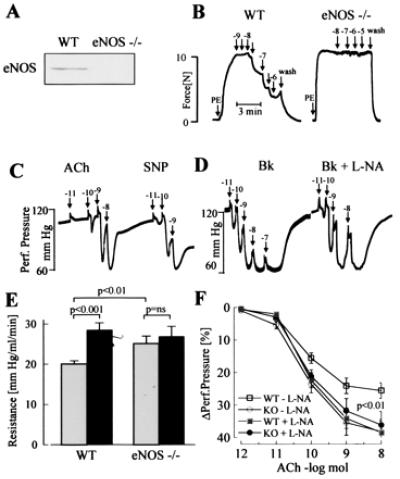
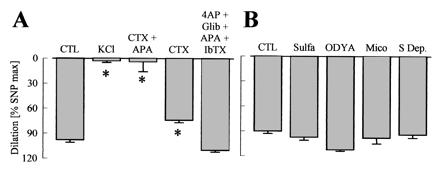
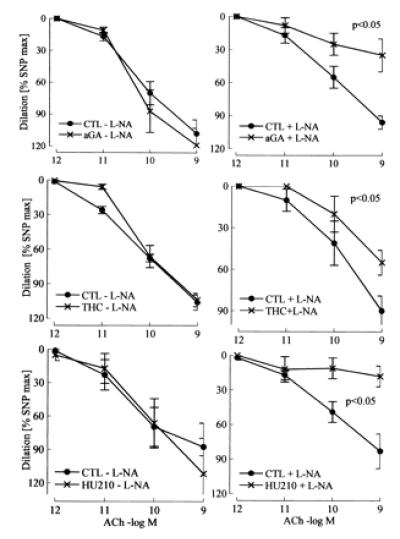
In addition to nitric oxide (NO) and prostacyclin (PGI(2)), the endothelium generates the endothelium-derived hyperpolarizing factor (EDHF). We set out to determine whether an EDHF-like response can be detected in wild-type (WT) and endothelial NO synthase knockout mice (eNOS -/-) mice. Vasodilator responses to endothelium-dependent agonists were determined in vivo and in vitro. In vivo, bradykinin induced a pronounced, dose-dependent decrease in mean arterial pressure (MAP) which did not differ between WT and eNOS -/- mice and was unaffected by treatment with N(omega)-nitro-l-arginine methyl ester and diclofenac. In the saline-perfused hindlimb of WT and eNOS -/- mice, marked N(omega)-nitro-l-arginine (l-NA, 300 micromol/liter)- and diclofenac-insensitive vasodilations in response to both bradykinin and acetylcholine (ACh) were observed, which were more pronounced than the agonist-induced vasodilation in the hindlimb of WT in the absence of l-NA. This endothelium-dependent, NO/PGI(2)-independent vasodilatation was sensitive to KCl (40 mM) and to the combination of apamin and charybdotoxin. Gap junction inhibitors (18alpha-glycyrrhetinic acid, octanol, heptanol) and CB-1 cannabinoid-receptor agonists (Delta(9)-tetrahydrocannabinol, HU210) impaired EDHF-mediated vasodilation, whereas inhibition of cytochrome P450 enzymes, soluble guanylyl cyclase, or adenosine receptors had no effect on EDHF-mediated responses. These results demonstrate that in murine resistance vessels the predominant agonist-induced endothelium-dependent vasodilation in vivo and in vitro is not mediated by NO, PGI(2), or a cytochrome P450 metabolite, but by an EDHF-like principle that requires functional gap junctions.
Figures
References
-
- Furchgott R F, Vanhoutte P M. FASEB J. 1989;3:2007–2018. - PubMed
-
- Furchgott R F. Biosci Rep. 1999;19:235–251. - PubMed
-
- Quilley J, Fulton D, McGiff J C. Biochem Pharmacol. 1997;54:1059–1170. - PubMed
-
- Cohen R A, Vanhoutte P M. Circulation. 1995;92:3337–3349. - PubMed
-
- Edwards G, Weston A H. Prog Drug Res. 1998;50:107–133. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
