

tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c
- PMID: 10950869
- PMCID: PMC316859
tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c
Abstract
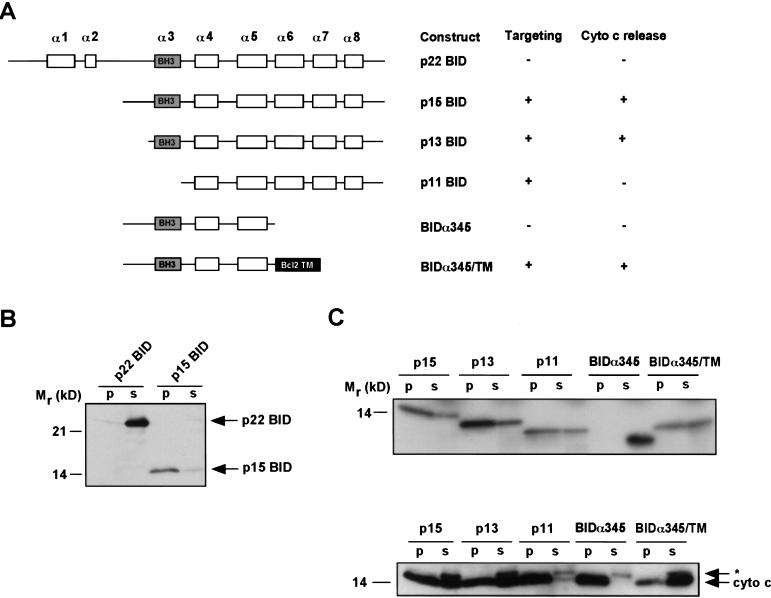
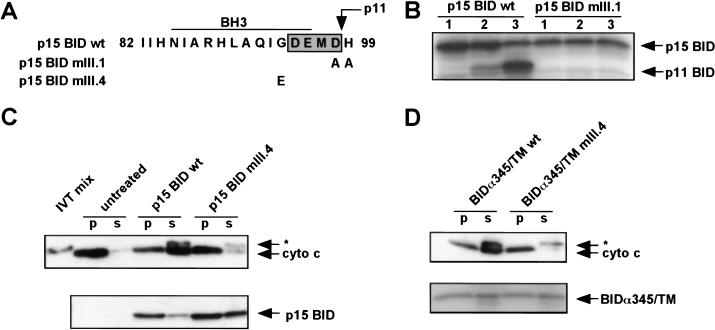
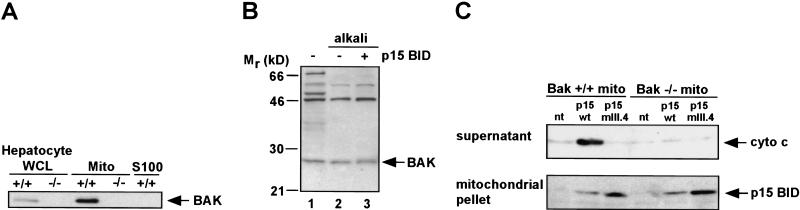
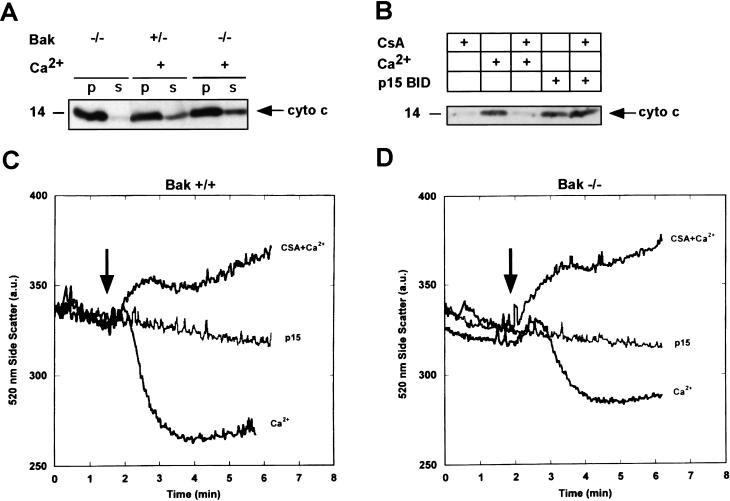
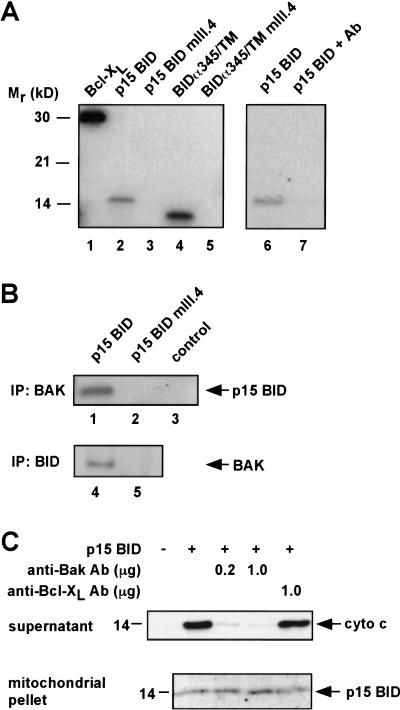
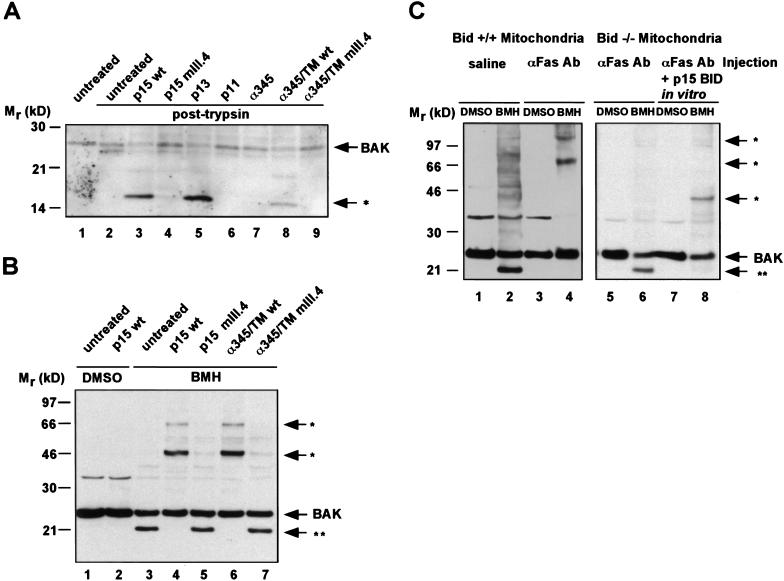
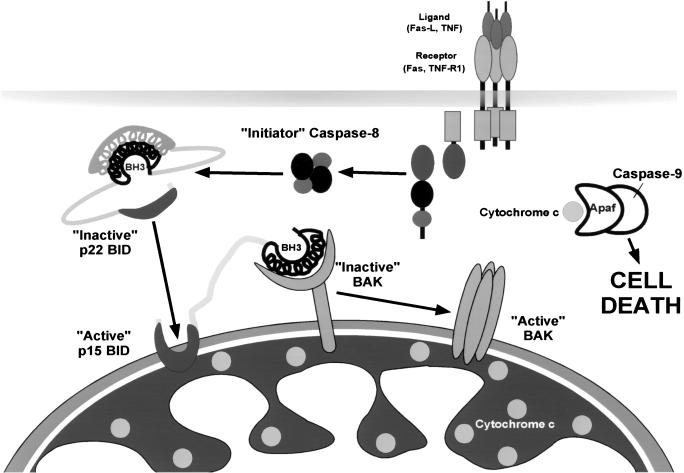
TNFR1/Fas engagement results in the cleavage of cytosolic BID to truncated tBID, which translocates to mitochondria. Immunodepletion and gene disruption indicate BID is required for cytochrome c release. Surprisingly, the three-dimensional structure of this BH3 domain-only molecule revealed two hydrophobic alpha-helices suggesting tBID itself might be a pore-forming protein. Instead, we demonstrate that tBID functions as a membrane-targeted death ligand in which an intact BH3 domain is required for cytochrome c release, but not for targeting. Bak-deficient mitochondria and blocking antibodies reveal tBID binds to its mitochondrial partner BAK to release cytochrome c, a process independent of permeability transition. Activated tBID results in an allosteric activation of BAK, inducing its intramembranous oligomerization into a proposed pore for cytochrome c efflux, integrating the pathway from death receptors to cell demise.
Figures
References
-
- Adams JM, Cory S. The Bcl-2 protein family: Arbiters of cell survival. Science. 1998;281:1322–1326. - PubMed
-
- Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277:370–372. - PubMed
-
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. - PubMed
-
- Chou JJ, Li H, Salvesen GS, Yuan J, Wagner G. Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell. 1999;96:615–624. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous