

Review
doi: 10.1172/JCI10830.
Insights into insulin resistance and type 2 diabetes from knockout mouse models
Affiliations
- PMID: 10953020
- PMCID: PMC380257
- DOI: 10.1172/JCI10830
Item in Clipboard
Review
Insights into insulin resistance and type 2 diabetes from knockout mouse models
J Clin Invest.
2000 Aug.
No abstract available
Figures
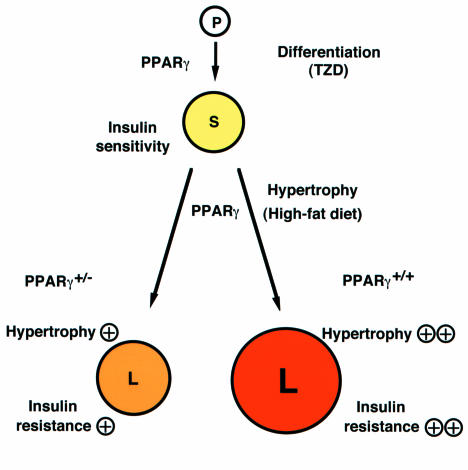
Roles of PPARγ in the adipocyte. In this model, PPARγ acts at two steps to regulate adipocyte size and insulin sensitivity. First, PPARγ promotes the differentiation of pre-adipocytes (P) to normal, insulin-sensitive, small adipocytes (S), a step that can be activated by providing artificial PPARγ agonists, the thiazolidinediones (TZD). PPARγ also plays a critical role in adipocyte hypertrophy and development of insulin resistance under a high-fat diet. In wild-type mice, such a diet promotes adipocyte hypertrophy, which converts small adipocytes (S) into large adipocytes (L). These latter cells, in turn, induce factors such as TNF-α and FFAs, which promote insulin resistance. PPARγ+/– animals therefore enjoy some protection from adipocyte hypertrophy and the development of insulin resistance under a high-fat diet. Adapted from ref. .
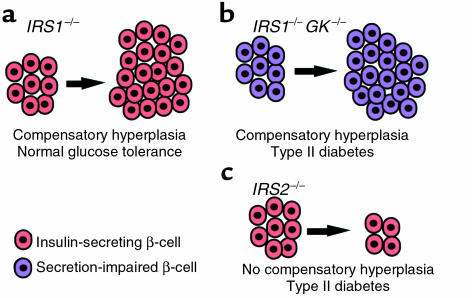
Three patterns of response of pancreatic β cells to insulin resistance. (a) In IRS–/– animals, glucose tolerance remains normal in the face of insulin resistance because their β cells (red) proliferate to compensate for reduced insulin responses. (b) In mice lacking both glucokinase and IRS-1 (GK–/–/IRS-1 double knockouts), a similar compensatory β-cell hyperplasia occurs, but the animals develop type 2 diabetes because their β cells (purple) are defective in glucose-stimulated insulin secretion. (c) IRS-2–/– mice, similarly, become overtly diabetic. These animals have functional β cells (red) that are unable to undergo hyperplasia to compensate for insulin resistance. Together, these results indicate that the compensatory response of β cells to insulin resistance plays a crucial role in preventing the development of type 2 diabetes. KO, knockout.
Comment in
-
Unraveling the mechanism of action of thiazolidinediones.J Clin Invest. 2000 Dec;106(11):1305-7. doi: 10.1172/JCI11705. J Clin Invest. 2000. PMID: 11104782 Free PMC article. No abstract available.
References
-
- Kadowaki T, et al. Risk factors for worsening to diabetes in subjects with impaired glucose tolerance. Diabetologia. 1984;26:44–49. - PubMed
-
- Taylor SI, Accili D, Imai Y. Insulin resistance or insulin deficiency: which is the primary cause of NIDDM? Diabetes. 1994;43:735–740. - PubMed
-
- Moller DE. Transgenic approaches to the pathogenesis of NIDDM. Diabetes. 1994;43:1394–1401. - PubMed
-
- Accili D, et al. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
