

Genome-wide detection of allelic imbalance using human SNPs and high-density DNA arrays
- PMID: 10958631
- PMCID: PMC2235196
- DOI: 10.1101/gr.10.8.1126
Genome-wide detection of allelic imbalance using human SNPs and high-density DNA arrays
Abstract
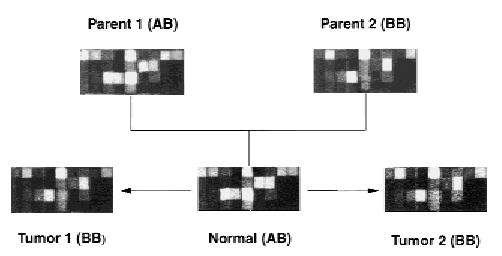
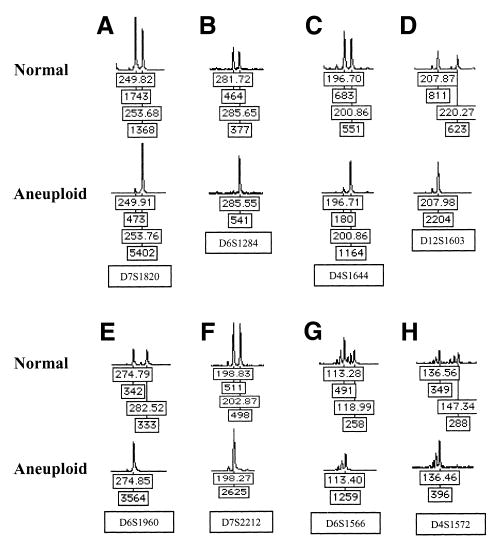
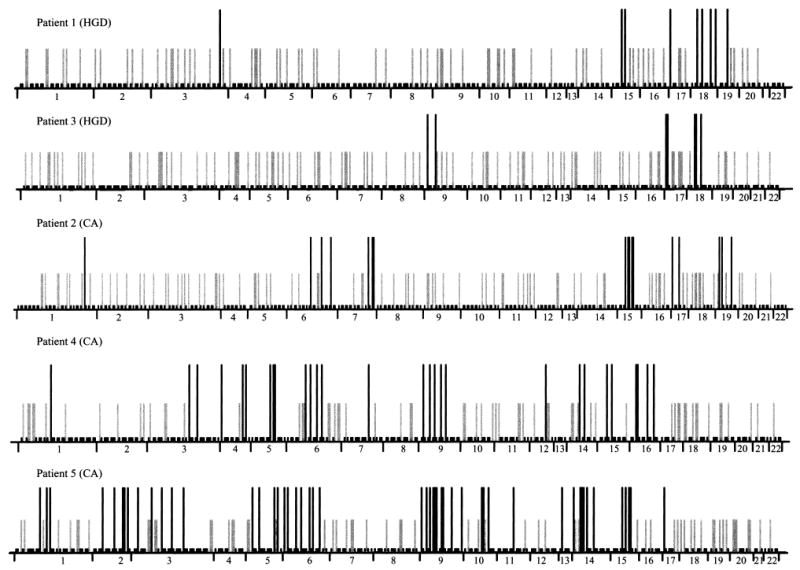
Most human cancers are characterized by genomic instability, the accumulation of multiple genetic alterations and allelic imbalance throughout the genome. Loss of heterozygosity (LOH) is a common form of allelic imbalance and the detection of LOH has been used to identify genomic regions that harbor tumor suppressor genes and to characterize tumor stages and progression. Here we describe the use of high-density oligonucleotide arrays for genome-wide scans for LOH and allelic imbalance in human tumors. The arrays contain redundant sets of probes for 600 genetic loci that are distributed across all human chromosomes. The arrays were used to detect allelic imbalance in two types of human tumors, and a subset of the results was confirmed using conventional gel-based methods. We also tested the ability to study heterogeneous cell populations and found that allelic imbalance can be detected in the presence of a substantial background of normal cells. The detection of LOH and other chromosomal changes using large numbers of single nucleotide polymorphism (SNP) markers should enable identification of patterns of allelic imbalance with potential prognostic and diagnostic utility.
Figures






References
-
- Baccichet A, Qualman SK, Sinnett D. Allelic loss in childhood acute lymphoblastic leukemia. Leuk Res. 1997;21:817–823. - PubMed
-
- Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ. Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene. 1996;12:1873–1878. - PubMed
-
- Boige V, Laurent-Puig P, Fouchet P, Flejou JF, Monges G, Bedossa P, Bioulac-Sage P, Capron F, Schmitz A, Olschwang S, et al. Concerted nonsyntenic allelic losses in hyperploid hepatocellular carcinoma as determined by a high-resolution allelotype. Cancer Res. 1997;57:1986–1990. - PubMed
-
- Brown MA. Tumor suppressor genes and human cancer. Adv Genet. 1997;36:45–135. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources