

Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals
- PMID: 10974035
- PMCID: PMC2193275
- DOI: 10.1084/jem.192.5.695
Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals
Abstract
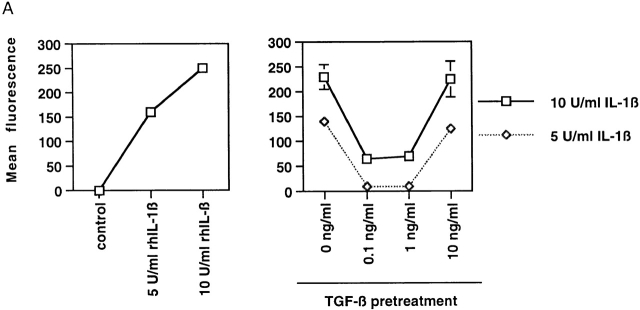
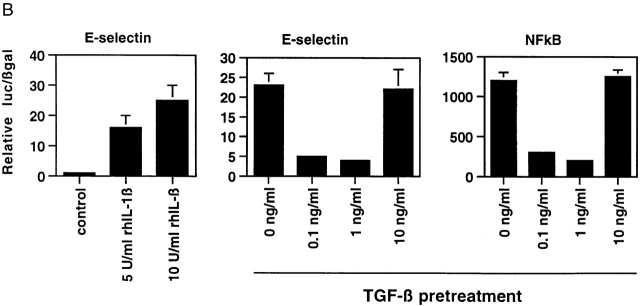
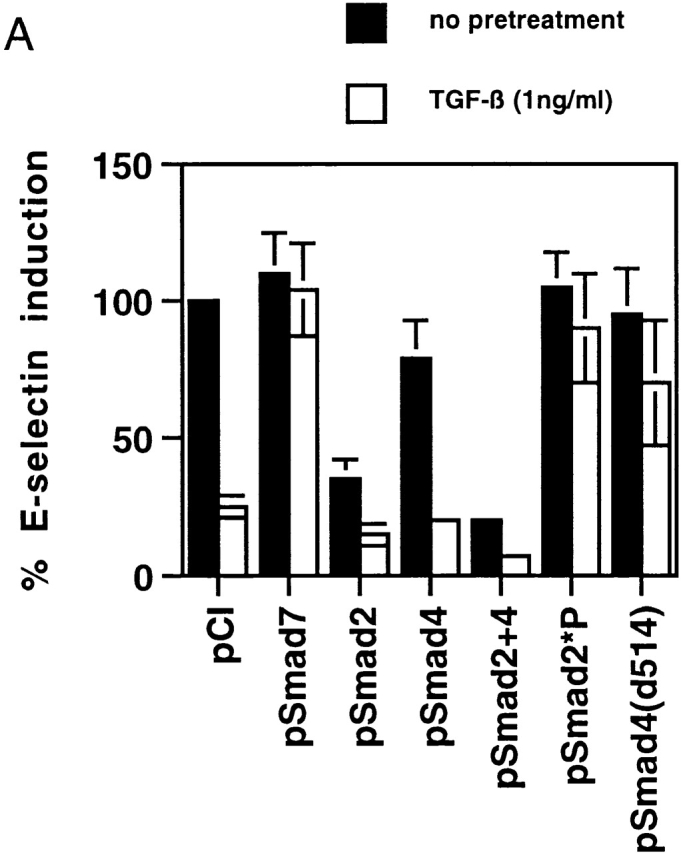
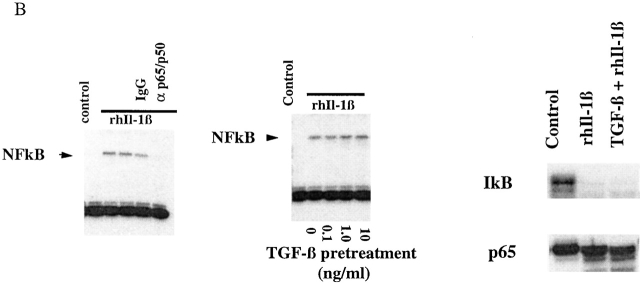
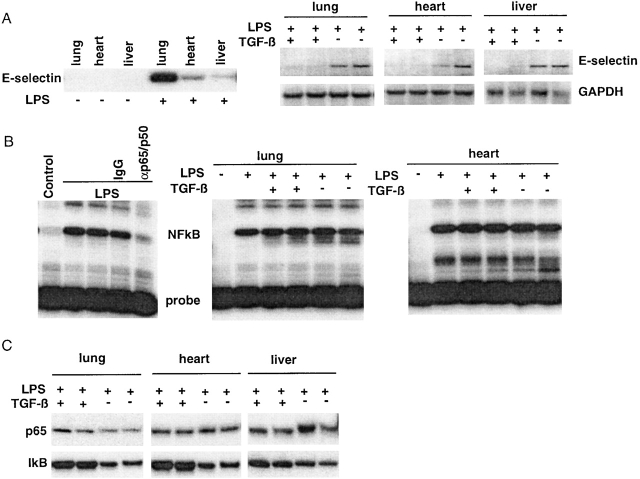
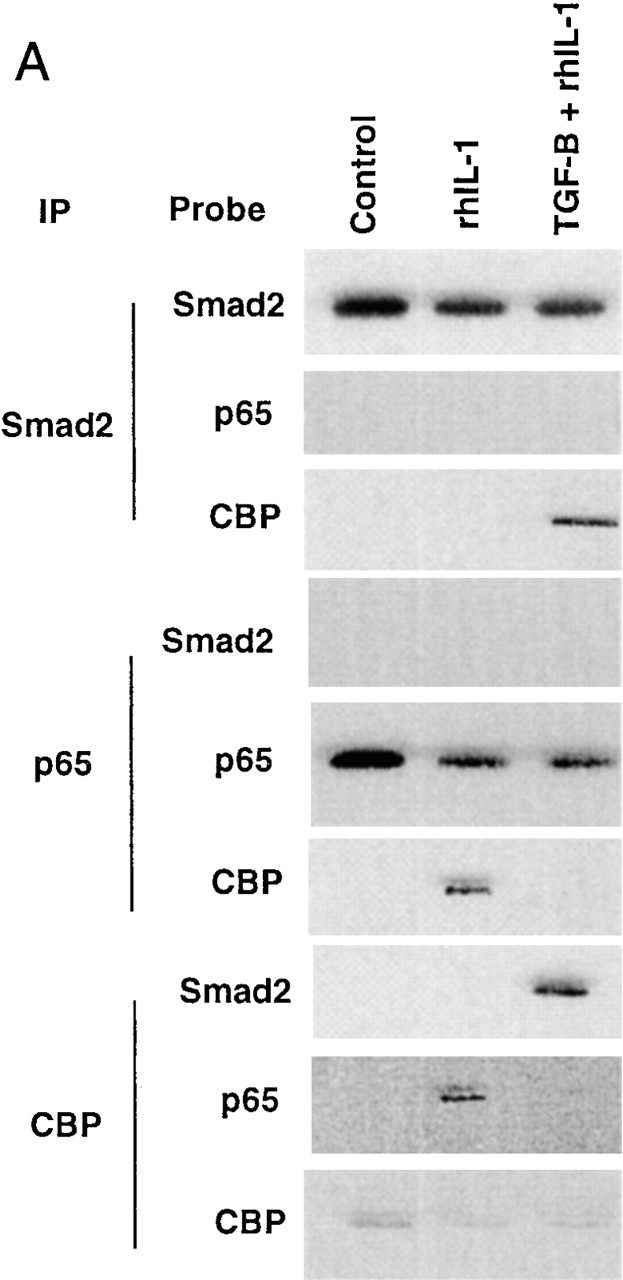
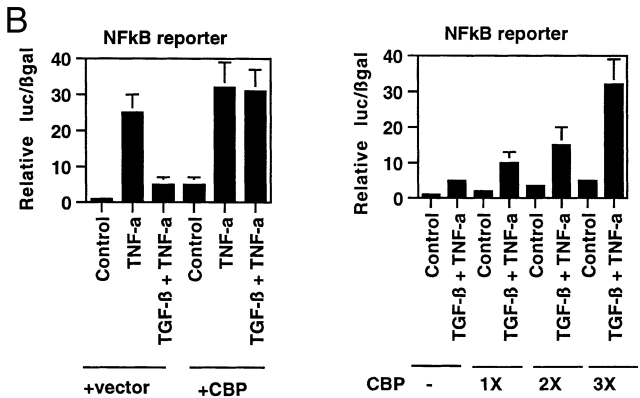
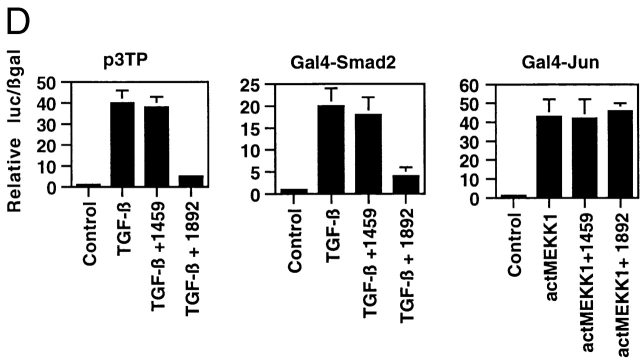
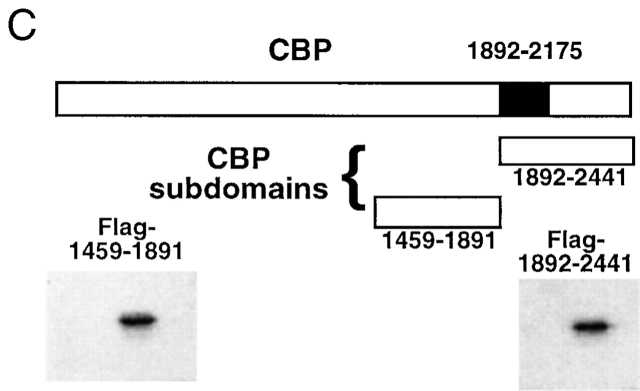
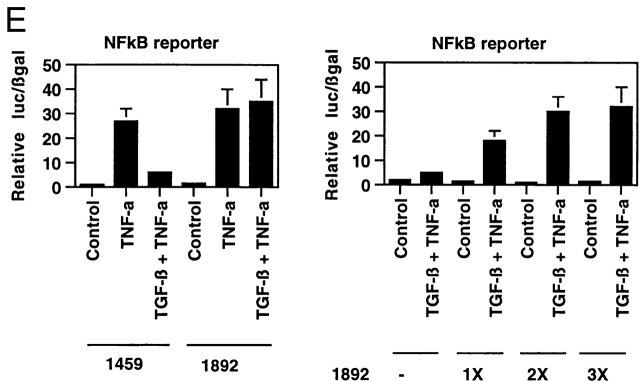
Transforming growth factor (TGF)-beta(1) is a pleiotropic cytokine/growth factor that is thought to play a critical role in the modulation of inflammatory events. We demonstrate that exogenous TGF-beta(1) can inhibit the expression of the proinflammatory adhesion molecule, E-selectin, in vascular endothelium exposed to inflammatory stimuli both in vitro and in vivo. This inhibitory effect occurs at the level of transcription of the E-selectin gene and is dependent on the action of Smad proteins, a class of intracellular signaling proteins involved in mediating the cellular effects of TGF-beta(1). Furthermore, we demonstrate that these Smad-mediated effects in endothelial cells result from a novel competitive interaction between Smad proteins activated by TGF-beta(1) and nuclear factor kappaB (NFkappaB) proteins activated by inflammatory stimuli (such as cytokines or bacterial lipopolysaccharide) that is mediated by the transcriptional coactivator cyclic AMP response element-binding protein (CREB)-binding protein (CBP). Augmentation of the limited amount of CBP present in endothelial cells (via overexpression) or selective disruption of Smad-CBP interactions (via a dominant negative strategy) effectively antagonizes the ability of TGF-beta(1) to block proinflammatory E-selectin expression. These data thus demonstrate a novel mechanism of interaction between TGF-beta(1)-regulated Smad proteins and NFkappaB proteins regulated by inflammatory stimuli in vascular endothelial cells. This type of signaling mechanism may play an important role in the immunomodulatory actions of this cytokine/growth factor in the cardiovascular system.
Figures
References
-
- Bevilacqua M.P., Stengelin S., Gimbrone M.A.J., Jr., Seed B. Endothelial leukocyte adhesion molecule 1an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–1165. - PubMed
-
- Collins T., Read M.A., Neish A.S., Whitley M.Z., Thanos D., Maniatis T. Transcriptional regulation of endothelial cell adhesion moleculesNF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. - PubMed
-
- Read M.A., Neish A.S., Luscinskas F.W., Palombella V.J., Maniatis T., Collins T. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity. 1995;2:493–506. - PubMed
-
- Read M.A., Whitley M.Z., Gupta S., Pierce J.W., Best J., Davis R.J., Collins T. Tumor necrosis factor alpha-induced E-selectin expression is activated by the nuclear factor-kappaB and c-JUN N-terminal kinase/p38 mitogen-activated protein kinase pathways. J. Biol. Chem. 1997;272:2753–2761. - PubMed
