

Changes in free and esterified cholesterol: hallmarks of acute renal tubular injury and acquired cytoresistance
- PMID: 10980139
- PMCID: PMC1885711
- DOI: 10.1016/S0002-9440(10)64613-5
Changes in free and esterified cholesterol: hallmarks of acute renal tubular injury and acquired cytoresistance
Abstract
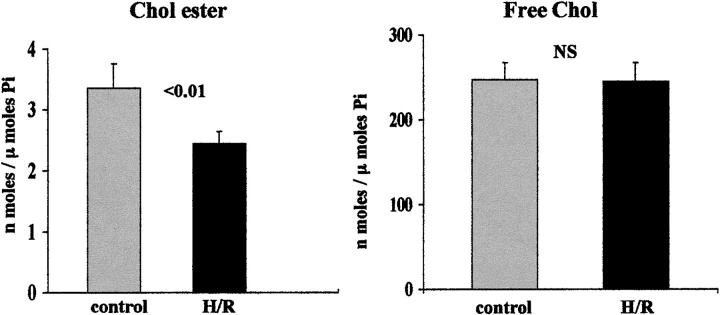
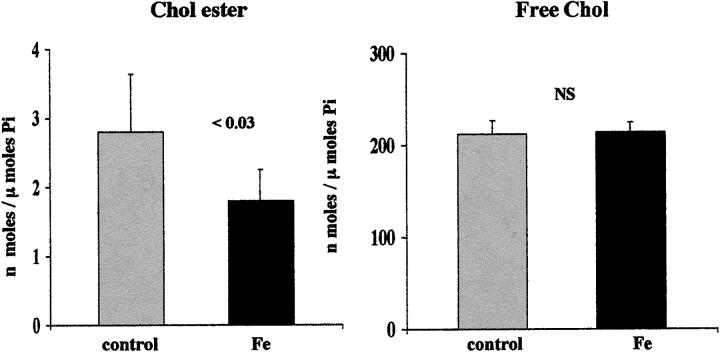
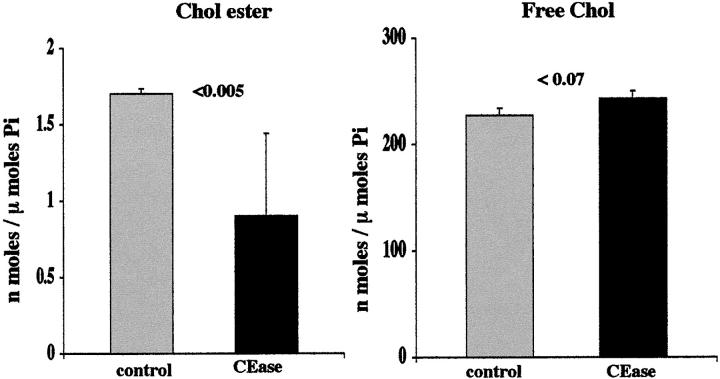
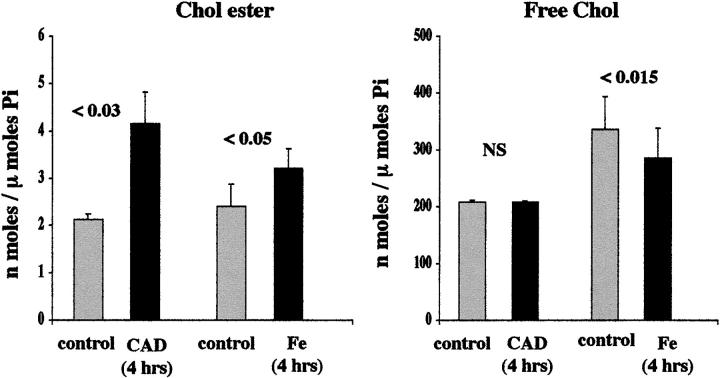
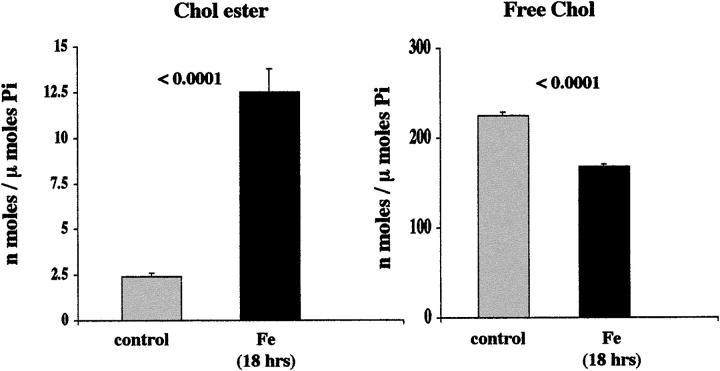
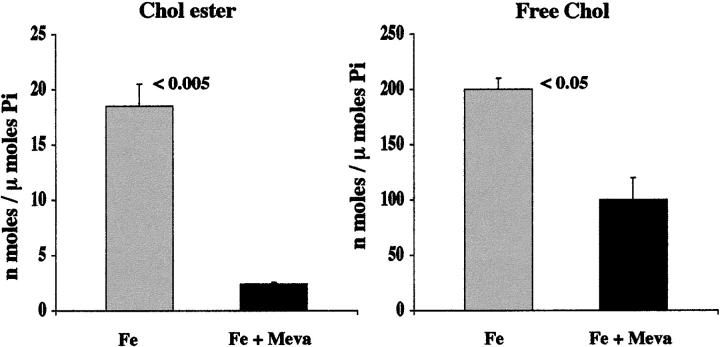
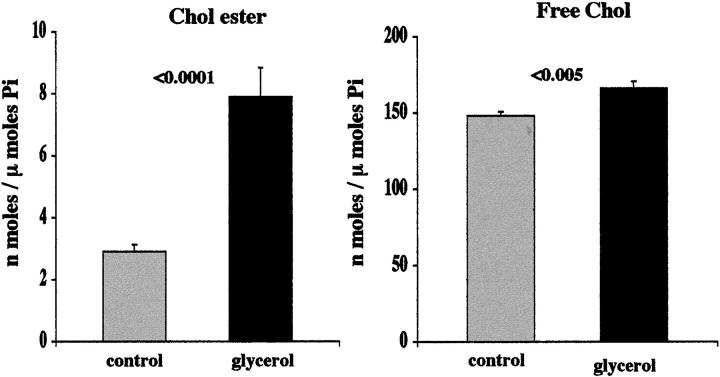
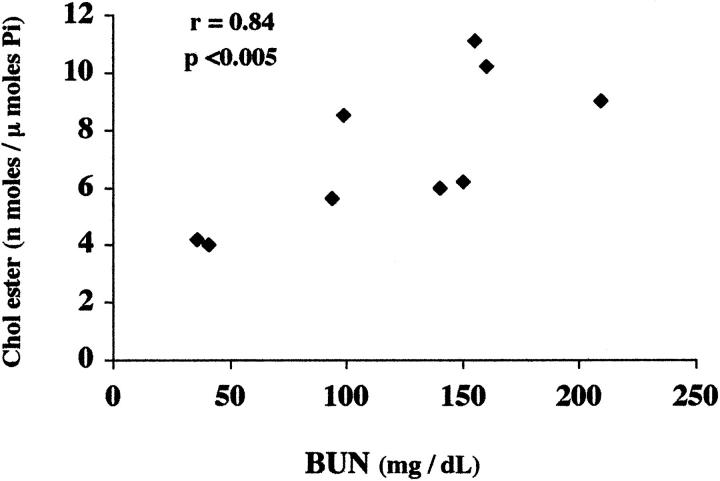
Acute tubular cell injury is accompanied by plasma membrane phospholipid breakdown. Although cholesterol is a dominant membrane lipid which interdigitates with, and impacts, phospholipid homeostasis, its fate during the induction and recovery phases of acute renal failure (ARF) has remained ill defined. The present study was performed to ascertain whether altered cholesterol expression is a hallmark of evolving tubular damage. Using gas chromatographic analysis, free cholesterol (FC) and esterified cholesterol (CE) were quantified in: 1) isolated mouse proximal tubule segments (PTS) after 30 minutes of hypoxic or oxidant (ferrous ammonium sulfate) injury; 2) cultured proximal tubule (HK-2) cells after 4 or 18 hours of either ATP depletion/Ca(2+) ionophore- or ferrous ammonium sulfate-mediated injury; and 3) in renal cortex 18 hours after induction of glycerol-induced myoglobinuric ARF, a time corresponding to the so-called "acquired cytoresistance" state (ie, resistance to further renal damage). Hypoxic and oxidant injury each induced approximately 33% decrements in CE (but not FC) levels in PTS, corresponding with lethal cell injury ( approximately 50 to 60% LDH release). When comparable CE declines were induced in normal PTS by exogenous cholesterol esterase treatment, proportionate lethal cell injury resulted. During models of slowly evolving HK-2 cell injury, progressive CE increments occurred: these were first noted at 4 hours, and reached approximately 600% by 18 hours. In vivo myoglobinuric ARF produced comparable renal cortical CE (and to a lesser extent FC) increments. Renal CE accumulation strikingly correlated with the severity of ARF (eg, blood urea nitrogen versus CE; r, 0.84). Mevastatin blocked cholesterol accumulation in injured HK-2 cells, indicating de novo synthesis was responsible. Acute tubule injury first lowers, then raises, tubule cholesterol content. Based on previous observations that cholesterol has cytoprotectant properties, the present findings have potential relevance for both the induction and maintenance phases of ARF.
Figures
References
-
- Bittman R: Has nature designed the cholesterol side chain for optimal interaction with phospholipids? Subcellular Biochemistry. 1997, :pp 145-171 vol 28, ch 6. Edited by R Bittman. New York, Plenum Press, Cholesterol: Its Functional and Metabolism in Biology and Medicine - PubMed
-
- Zager RA: Increased proximal tubular cholesterol content: implications for cell injury and “acquired cytoresistance.” Kidney Int 1999, 56:1788–1797 - PubMed
-
- Presti FT: The role of cholesterol in regulating membrane fluidity. Aloia RC Boggs JM eds. Membrane Fluidity in Biology, Cellular Aspects, 1985, vol 4.:pp 97-146 Academic Press, New York
-
- Presti FT, Pace RJ, Chan SI: Cholesterol-phospholipid interaction in membranes. 2. Stoichiometry and molecular packing of cholesterol-rich domains. Biochemistry 1982, 21:3831-3835 - PubMed
-
- Bittman R: Sterol exchange between mycoplasma membranes and vesicles. Yeagle PL eds. Biology of Cholesterol. 1988, :pp 173-195 CRC Press, Boca Raton
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
