

Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen Gly-X-Y triple helix
- PMID: 11007540
- PMCID: PMC1288550
- DOI: 10.1016/S0002-9297(07)62938-3
Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen Gly-X-Y triple helix
Abstract
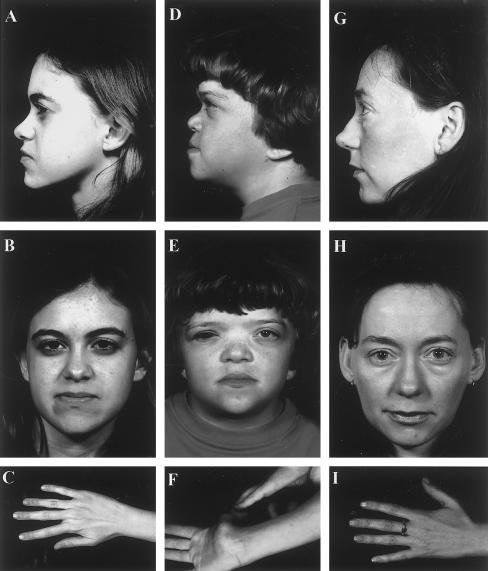
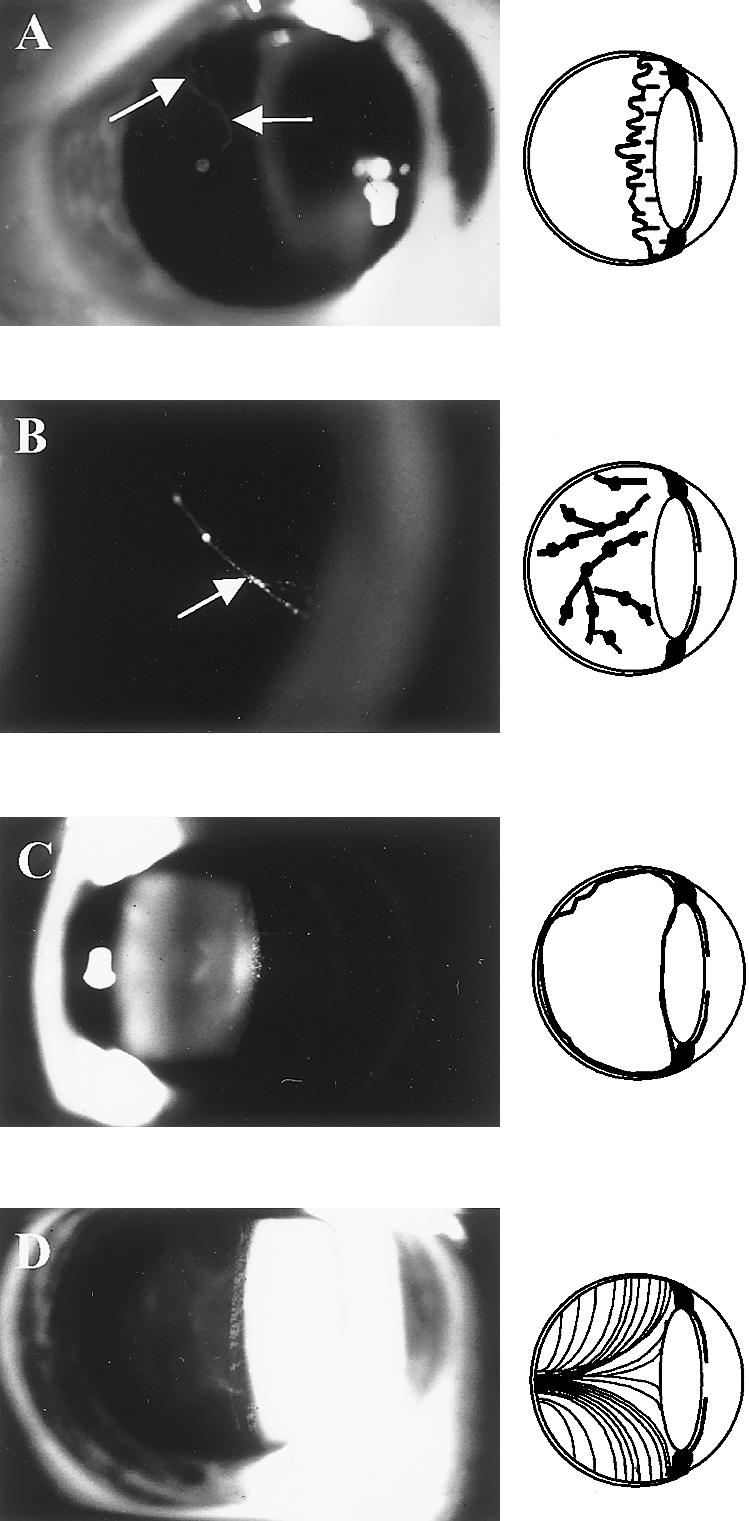
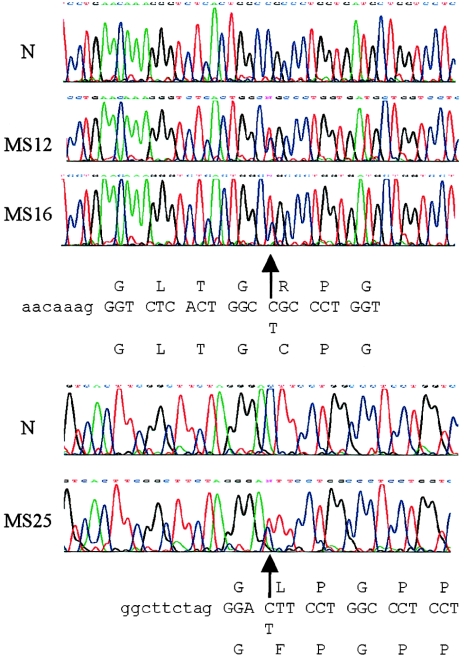
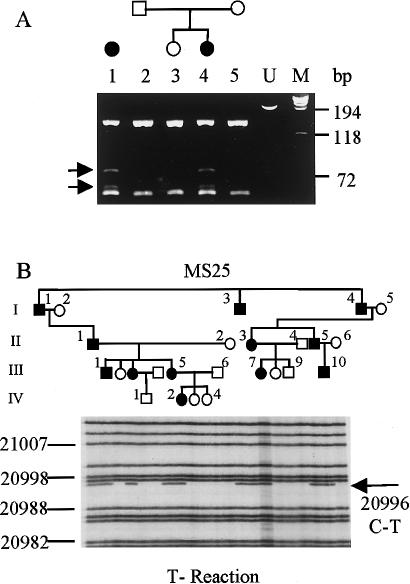
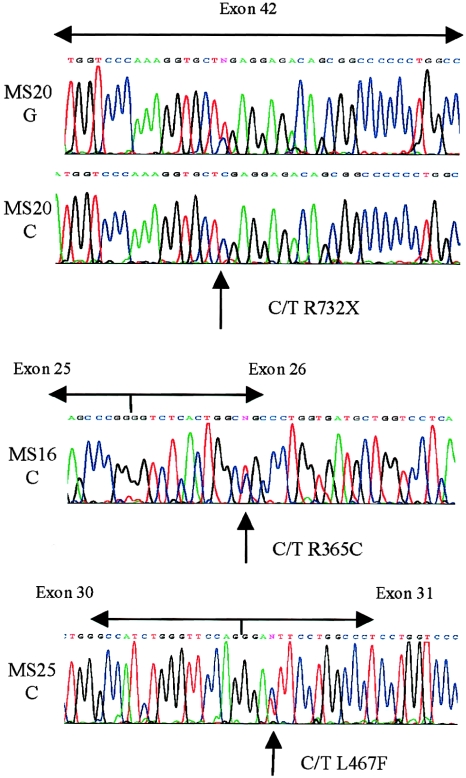
Stickler syndrome is a dominantly inherited disorder characterized by arthropathy, midline clefting, hearing loss, midfacial hypoplasia, myopia, and retinal detachment. These features are highly variable both between and within families. Mutations causing the disorder have been found in the COL2A1 and COL11A1 genes. Premature termination codons in COL2A1 that result in haploinsufficiency of type II collagen are a common finding. These produce a characteristic congenital "membranous" anomaly of the vitreous of all affected individuals. Experience has shown that vitreous slit-lamp biomicroscopy can distinguish between patients with COL2A1 mutations and those with dominant negative mutations in COL11A1, who produce a different "beaded" vitreous phenotype. Here we characterize novel dominant negative mutations in COL2A1 that result in Stickler syndrome. Both alter amino acids in the X position of the Gly-X-Y triple-helical region. A recurrent R365C mutation occurred in two unrelated sporadic cases and resulted in the membranous vitreous anomaly associated with haploinsufficiency. In a large family with linkage to COL2A1, with a LOD score of 2.8, a unique L467F mutation produced a novel "afibrillar" vitreous gel devoid of all normal lamella structure. These data extend the mutation spectrum of the COL2A1 gene and help explain the basis for the different vitreous phenotypes seen in Stickler syndrome.
Figures
References
Electronic-Database Information
-
- Genome Database, The, http://gdbwww.gdb.org (for COL2A1 [accession number L10347])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for Marshall syndrome [MIM 154780] and Stickler syndrome [MIM 108300, MIM 184840, and MIM 120280])
References
-
- Anderson DW, Tromp G, Kuivaniemi H, Ricketts M, Pope FM, Stolle CA, Boyd CD (1994) A type III procollagen gene mutation in a patient with late onset aneurysms. Matrix Biol 14:392
-
- Annunen S, Korkko J, Czarny M, Warman ML, Brunner HG, Kaariainen H, Mullikan JB, Tranebjarg L, Brooks DG, Cox GF, Cruysberg JR, Curtis MA, Davenport SL, Friedrich CA, Kaitila I, Krawczynski MR, Latos-Bielenska A, Mukai S, Olsen BR, Shinno N, Somer M, Vikkula M, Zlotogora J, Prockop DJ, Ala-Kokko L (1999) Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet 65: 974–983 - PMC - PubMed
-
- Ballo R, Beighton PH, Ramesar RS (1998) Stickler-like syndrome due to a dominant negative mutation in the COL2A1 gene. Am J Med Genet 80:6–11 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
