

Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism
- PMID: 11013134
- PMCID: PMC1288551
- DOI: 10.1086/303105
Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism
Abstract
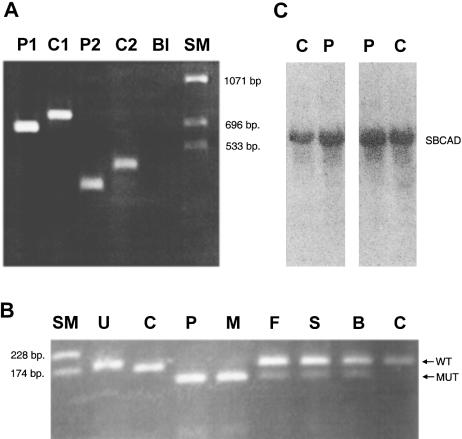
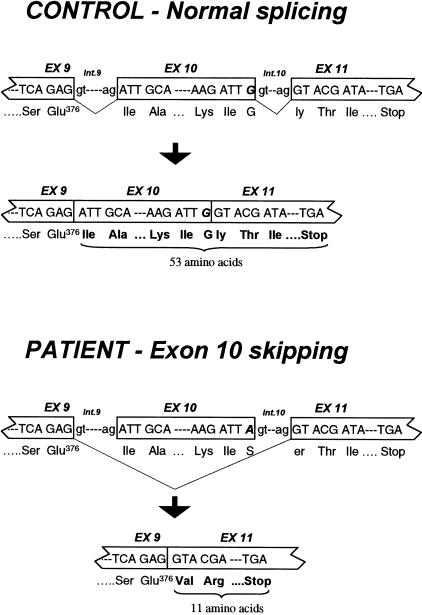
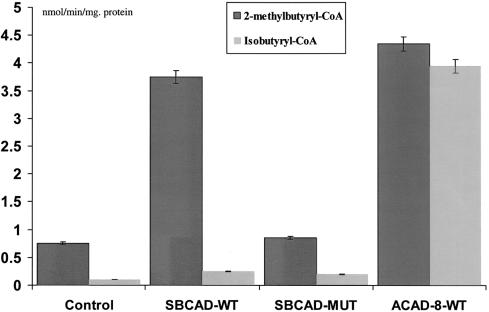
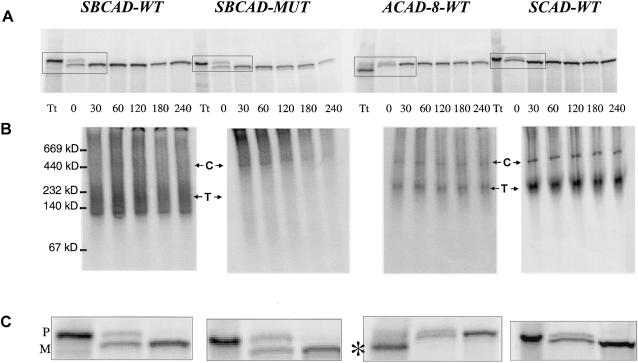
Acyl-CoA dehydrogenase (ACAD) defects in isoleucine and valine catabolism have been proposed in clinically diverse patients with an abnormal pattern of metabolites in their urine, but they have not been proved enzymatically or genetically, and it is unknown whether one or two ACADs are involved. We investigated a patient with isolated 2-methylbutyrylglycinuria, suggestive of a defect in isoleucine catabolism. Enzyme assay of the patient's fibroblasts, using 2-methylbutyryl-CoA as substrate, confirmed the defect. Sequence analysis of candidate ACADs revealed heterozygosity for the common short-chain ACAD A625 variant allele and no mutations in ACAD-8 but a 100-bp deletion in short/branched-chain ACAD (SBCAD) cDNA from the patient. Our identification of the SBCAD gene structure (11 exons; >20 kb) enabled analysis of genomic DNA. This showed that the deletion was caused by skipping of exon 10, because of homozygosity for a 1228G-->A mutation in the patient. This mutation was not present in 118 control chromosomes. In vitro transcription/translation experiments and overexpression in COS cells confirmed the disease-causing nature of the mutant SBCAD protein and showed that ACAD-8 is an isobutyryl-CoA dehydrogenase and that both wild-type proteins are imported into mitochondria and form tetramers. In conclusion, we report the first mutation in the SBCAD gene, show that it results in an isolated defect in isoleucine catabolism, and indicate that ACAD-8 is a mitochondrial enzyme that functions in valine catabolism.
Figures
References
Electronic-Database Information
-
- Genbank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for human SBCAD gene [accession numbers AF260668–AF260678])
References
-
- Amir N, Elpeleg ON, Shalev RS, Christensen E (1989) Glutaric aciduria type I: enzymatic and neuroradiologic investigations of two kindreds. J Pediatr 114:98–99 - PubMed
-
- Andresen BS, Bross P, Udvari S, Kirk J, Gray RGF, Kmock S, Chamoles N, Knudsen I, Winter V, Wilcken B, Yokota I, Hart K, Packman S, Harpey JP, Saudubray JM, Hale DE, Bolund L, Kolvraa S, Gregersen N (1997) The molecular basis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency in compound heterozygous patients: is there a correlation between genotype and phenotype? Hum Mol Genet 6:695–708 - PubMed
-
- Andresen BS, Olpin S, Poorthuis B, Scholte HR, Vianey-Saban C, Wanders RJA, Ijlst L, Morris A, Pourfarzam M, Bartlett K, Baumgartner ER, deKlerk JB, Schroeder LD, Corydon TJ, Lund H, Winter V, Bross P, Bolund L, Gregersen N (1999) It is possible to correlate genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase (VLCAD) deficiency. Am J Hum Genet 64:479–494 - PMC - PubMed
-
- Binzak B, Willard J, Vockley J (1998) Identification of the catalytic residue of human short/branched chain acyl-CoA dehydrogenase by in vitro mutagenesis. Biochim Biophys Acta 1382:137–142 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
