

Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta
- PMID: 11013220
- PMCID: PMC302105
- DOI: 10.1093/emboj/19.19.5178
Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15(Ink4B) transcription in response to TGF-beta
Abstract
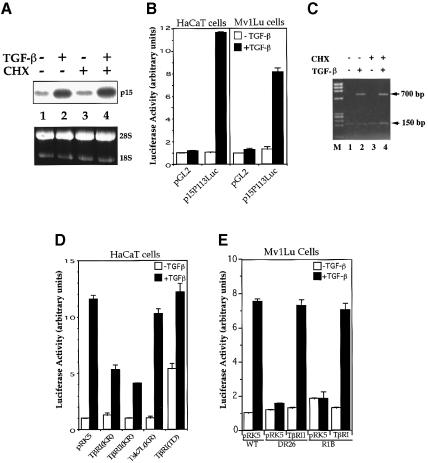
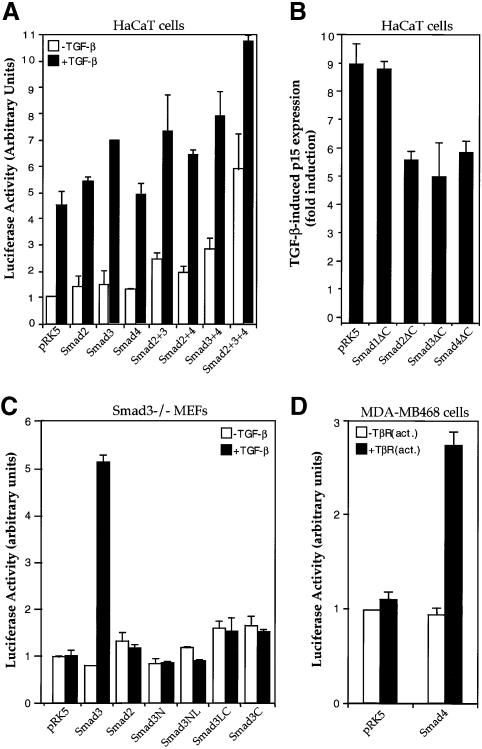
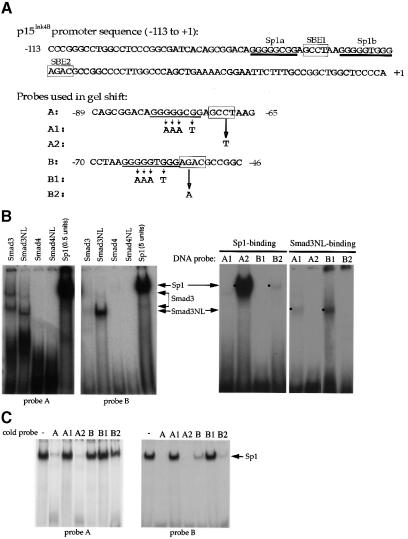
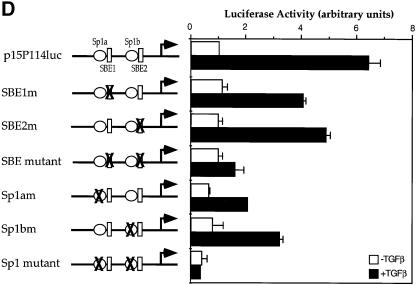
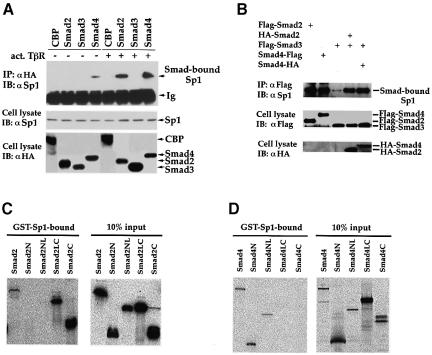
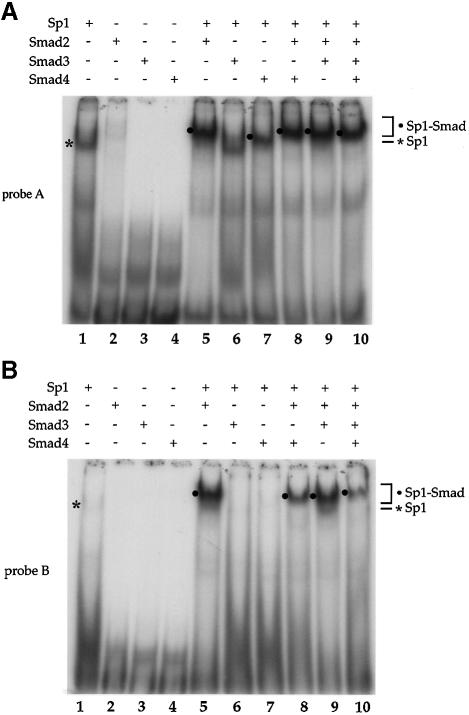
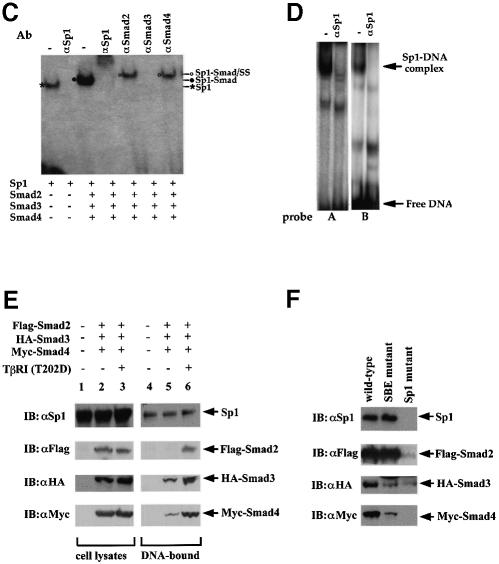
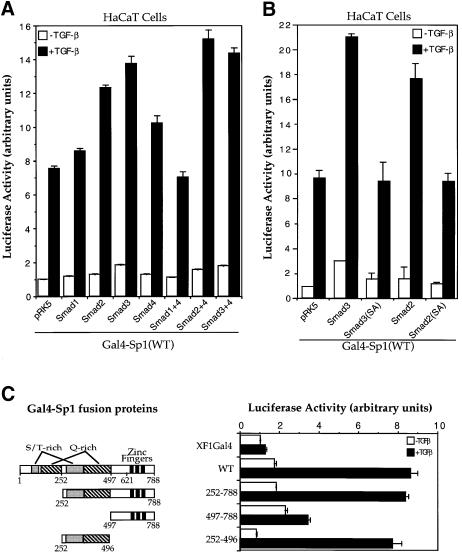
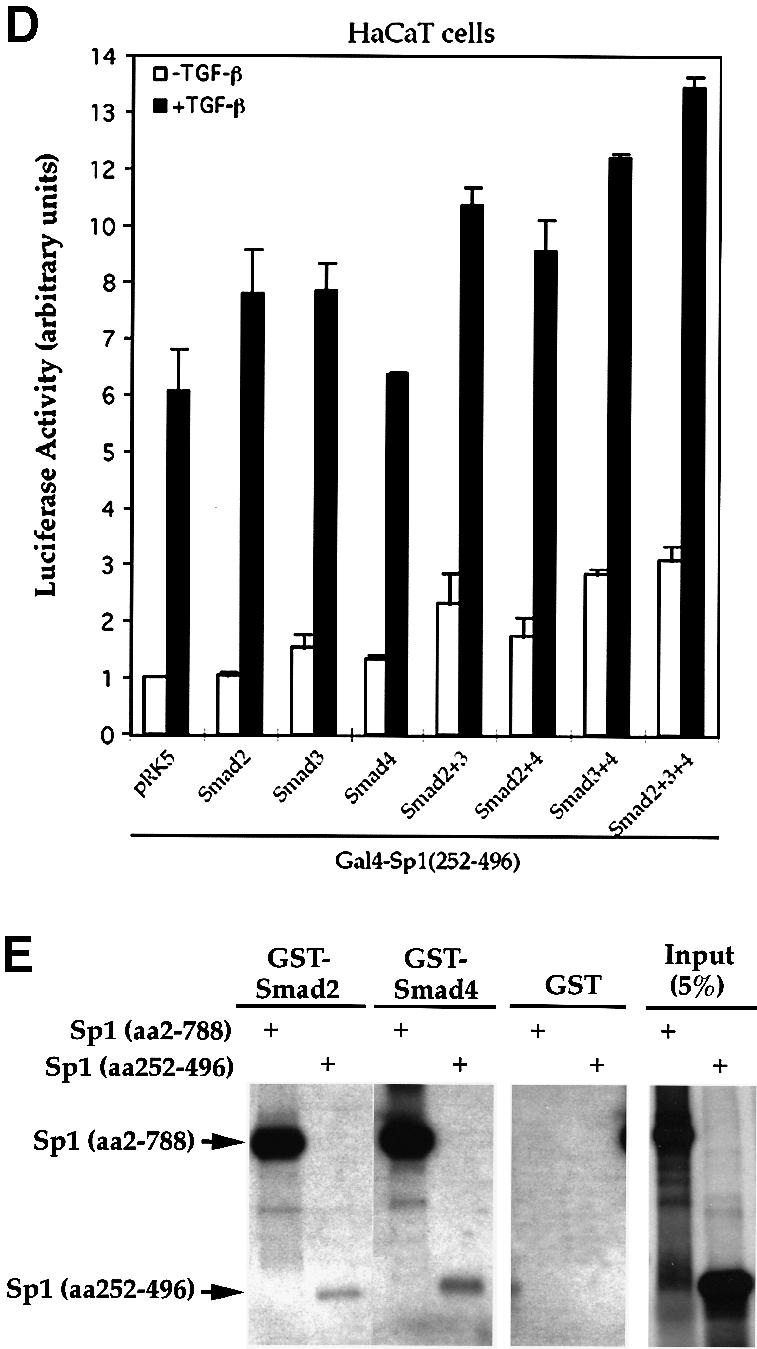
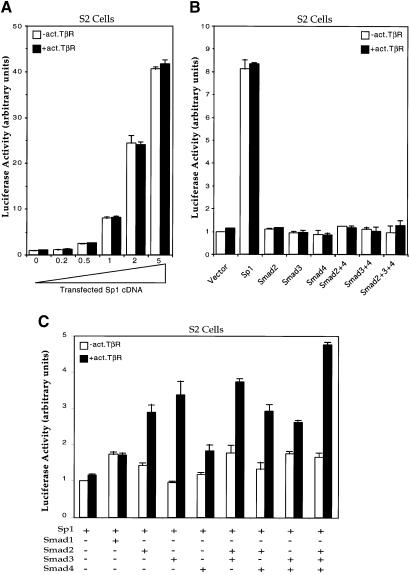
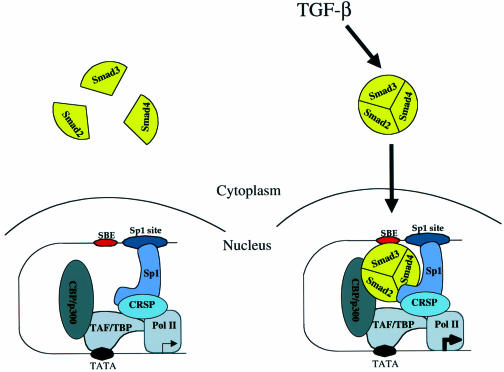
Transforming growth factor-beta (TGF-beta) arrests growth of epithelial cells by inducing the transcription of p15(Ink4B), a cyclin-dependent kinase inhibitor. In this study, we demonstrate that p15(Ink4B) induction was mediated by a TGF-beta-induced complex of Smad2, Smad3, Smad4 and Sp1. Mutations in the Sp1- or Smad-binding sequences decreased or abolished the TGF-beta responsiveness of the p15(Ink4B) promoter. Interference with, or deficiency in, Smad2, Smad3 or Smad4 functions also reduced or abolished the TGF-beta-dependent p15(Ink4B) induction, whereas the absence of Sp1 reduced the basal and TGF-beta-induced p15(Ink4B) transcription. In the nucleoprotein complex, Smad2 interacted through its C-domain with Sp1 and enhanced the DNA binding and transcriptional activity of Sp1. Smad3 interacted indirectly with Sp1 through its association with Smad2 and/or Smad4, and bound directly to the p15(Ink4B) promoter. Finally, Smad4 interacted through its N-domain with Sp1. Our data demonstrate the physical interactions and functional cooperativity of Sp1 with a complex of Smad2, Smad3 and Smad4 in the induction of the p15(Ink4B) gene. These findings explain the tumor suppressor roles of Smad2 and Smad4 in growth arrest signaling by TGF-beta.
Figures
References
-
- Abdollah S., Macías-Silva,M., Tsukazaki,T., Hayashi,H., Attisano,L. and Wrana,J.L. (1997) TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2–Smad4 complex formation and signaling. J. Biol. Chem., 272, 27678–27685. - PubMed
-
- Arrick B.A. and Derynck,R. (1996) The biological role of transforming growth factor-β in cancer development. In Waxman,J. (ed.), Molecular Endocrinology of Cancer. Cambridge University Press, Cambridge, UK, pp. 51–78.
-
- Attisano L. and Wrana,J.L. (1998) Mads and Smads in TGFβ signalling. Curr. Opin. Cell Biol., 10, 188–194. - PubMed
-
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1998) Current Protocols in Molecular Biology. John Wiley and Sons, Inc., New York, NY.
-
- Bae H.W., Geiser,A.G., Kim,D.H., Chung,M.T., Burmester,J.K., Sporn,M.B., Roberts,A.B. and Kim,S.-J. (1995) Characterization of the promoter region of the human transforming growth factor-β type II receptor gene. J. Biol. Chem., 270, 29460–29468. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
