

Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes
- PMID: 11027285
- PMCID: PMC86425
- DOI: 10.1128/MCB.20.21.8157-8167.2000
Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes
Abstract
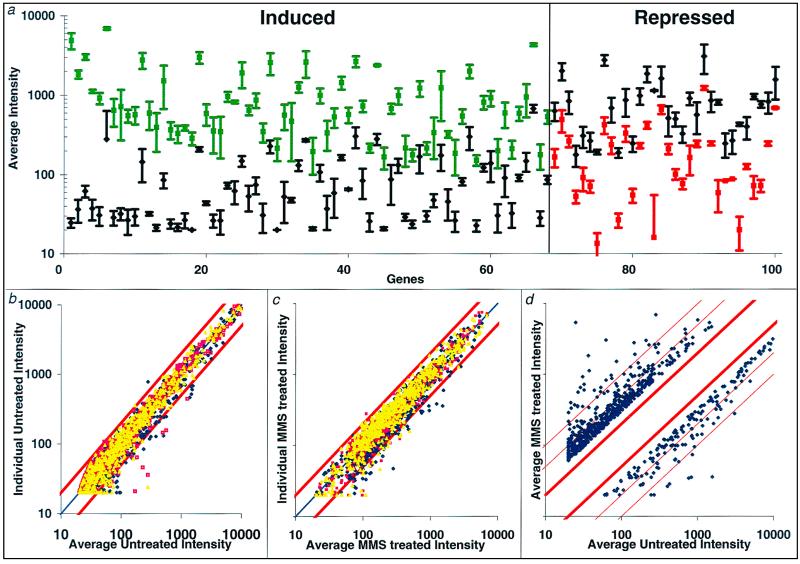
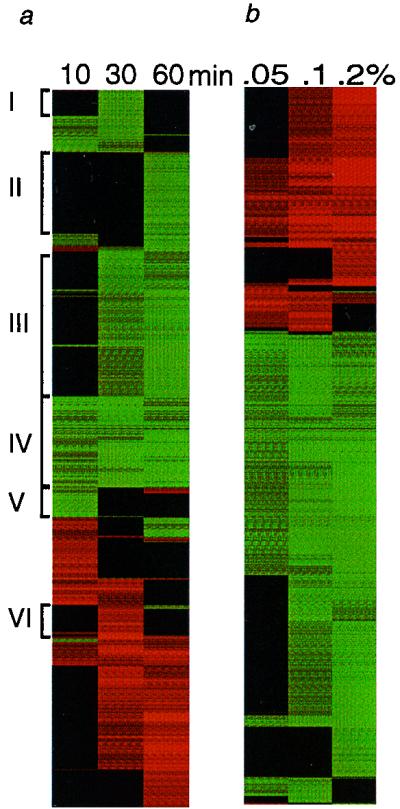
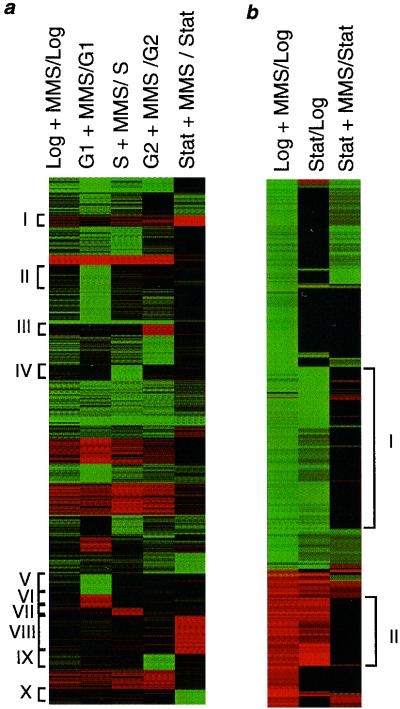
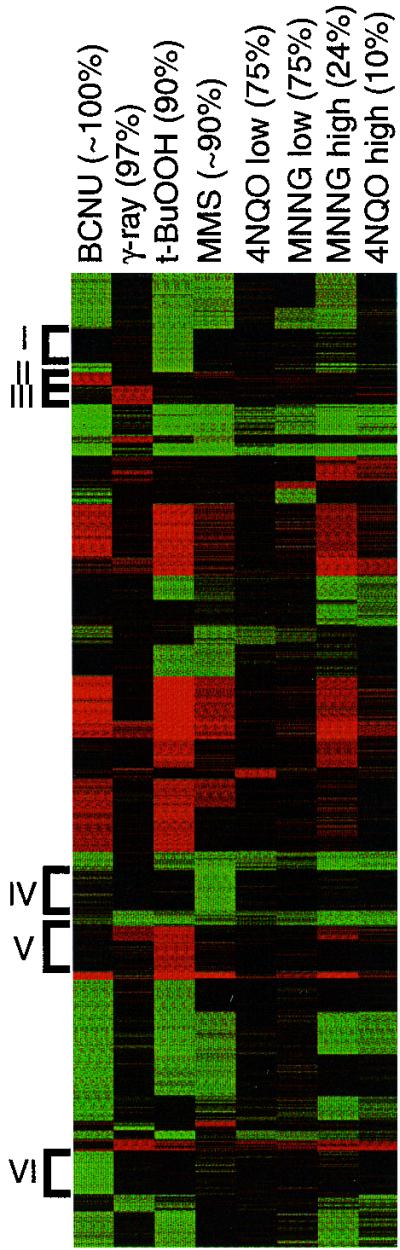
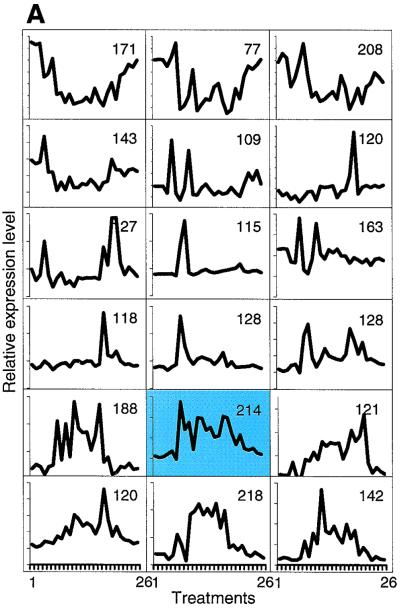
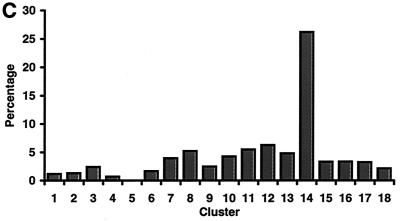
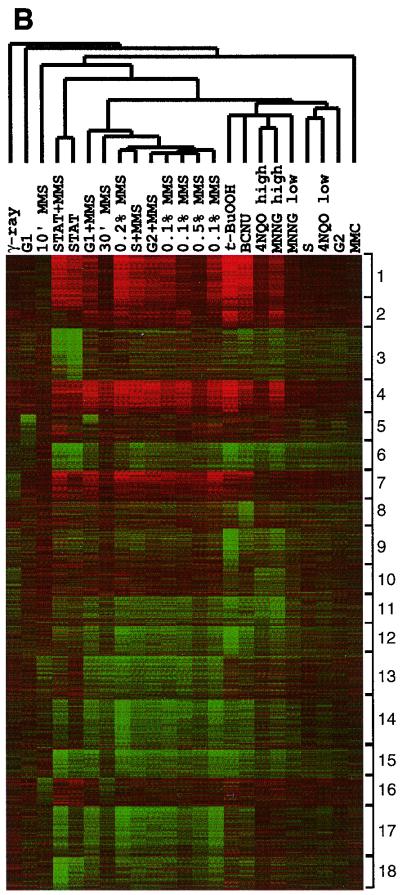
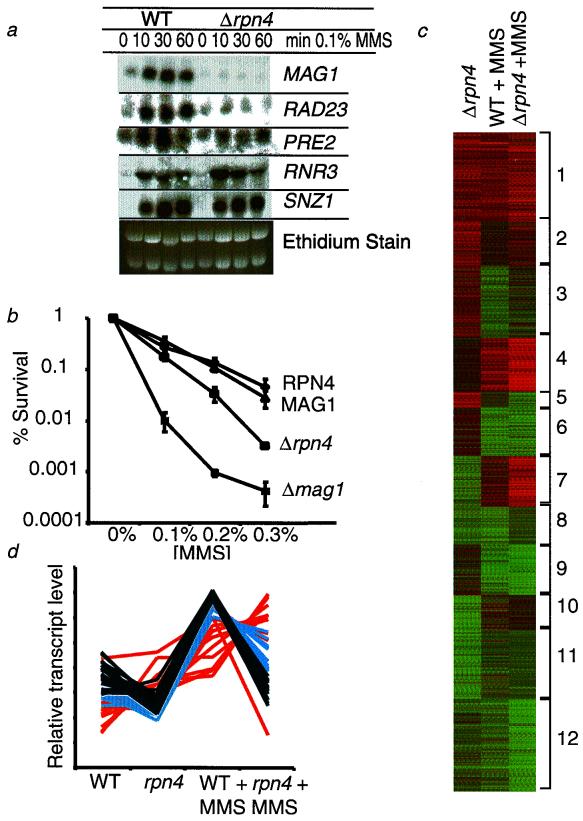
Exposure to carcinogenic alkylating agents, oxidizing agents, and ionizing radiation modulates transcript levels for over one third of Saccharomyces cerevisiae's 6,200 genes. Computational analysis delineates groups of coregulated genes whose upstream regions bear known and novel regulatory sequence motifs. One group of coregulated genes contain a number of DNA excision repair genes (including the MAG1 3-methyladenine DNA glycosylase gene) and a large selection of protein degradation genes. Moreover, transcription of these genes is modulated by the proteasome-associated protein Rpn4, most likely via its binding to MAG1 upstream repressor sequence 2-like elements, that turn out to be almost identical to the recently identified proteasome-associated control element (G. Mannhaupt, R. Schnall, V. Karpov, I. Vetter, and H. Feldmann, FEBS Lett. 450:27-34, 1999). We have identified a large number of genes whose transcription is influenced by Rpn4p.
Figures
References
-
- Ball C A, Dolinski K, Dwight S S, Harris M A, Issel-Tarver L, Kasarskis A, Scafe C R, Sherlock G, Binkley G, Jin H, Kaloper M, Orr S D, Schroeder M, Weng S, Zhu Y, Botstein D, Cherry J M. Integrating functional genomic information into the Saccharomyces genome database. Nucleic Acids Res. 2000;28:77–80. - PMC - PubMed
-
- Cho R J, Campbell M J, Winzeler E A, Steinmetz L, Conway A, Wodicka L, Wolfsberg T G, Gabrielian A E, Landsman D, Lockhart D J, Davis R W. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol Cell. 1998;2:65–73. - PubMed
-
- Chu S, DeRisi J, Elsen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases