

PERK mediates cell-cycle exit during the mammalian unfolded protein response
- PMID: 11035797
- PMCID: PMC18814
- DOI: 10.1073/pnas.220247197
PERK mediates cell-cycle exit during the mammalian unfolded protein response
Abstract
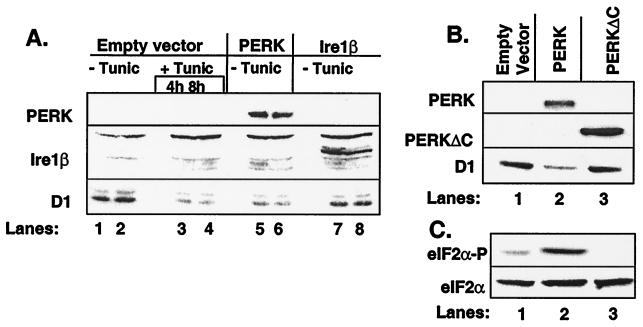
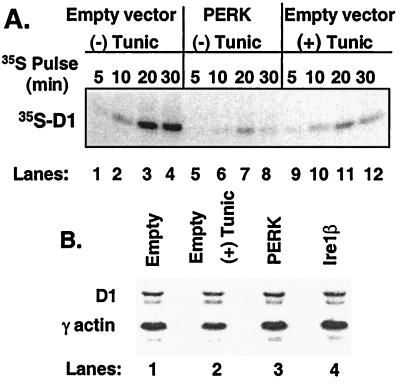
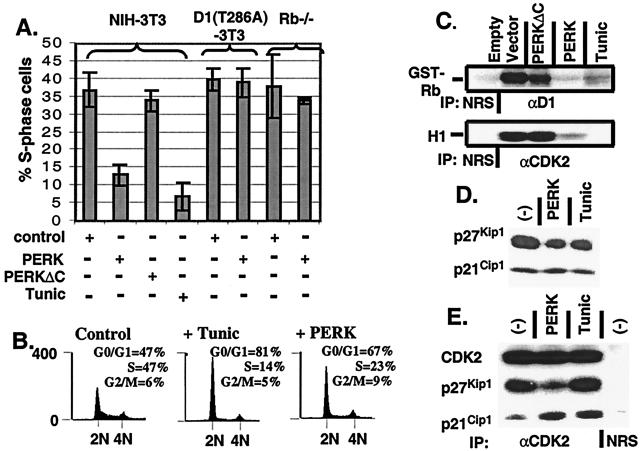
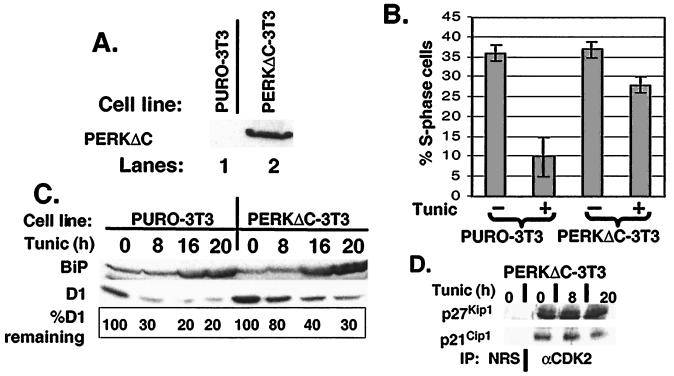
The accumulation of unfolded proteins in the endoplasmic reticulum (ER) triggers the unfolded protein response (UPR)-signaling pathway. The UPR coordinates the induction of ER chaperones with decreased protein synthesis and growth arrest in the G(1) phase of the cell cycle. Three ER transmembrane protein kinases (Ire1alpha, Ire1beta, and PERK) have been implicated as proximal effectors of the mammalian UPR. We now demonstrate that activation of PERK signals the loss of cyclin D1 during the UPR, culminating in cell-cycle arrest. Overexpression of wild-type PERK inhibited cyclin D1 synthesis in the absence of ER stress, thereby inducing a G(1) phase arrest. PERK expression was associated with increased phosphorylation of the translation elongation initiation factor 2alpha (eIF2alpha), an event previously shown to block cyclin D1 translation. Conversely, a truncated form of PERK lacking its kinase domain acted as a dominant negative when overexpressed in cells, attenuating both cyclin D1 loss and cell-cycle arrest during the UPR without compromising induction of ER chaperones. These data demonstrate that PERK serves as a critical effector of UPR-induced growth arrest, linking stress in the ER to control of cell-cycle progression.
Figures
Comment in
-
Pausing to decide.Proc Natl Acad Sci U S A. 2000 Nov 7;97(23):12396-7. doi: 10.1073/pnas.250476097. Proc Natl Acad Sci U S A. 2000. PMID: 11058174 Free PMC article. Review. No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
