

Testing the hypothesis of a recombinant origin of human immunodeficiency virus type 1 subtype E
- PMID: 11044120
- PMCID: PMC110950
- DOI: 10.1128/jvi.74.22.10752-10765.2000
Testing the hypothesis of a recombinant origin of human immunodeficiency virus type 1 subtype E
Abstract
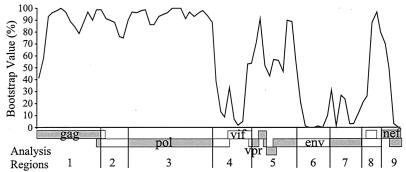
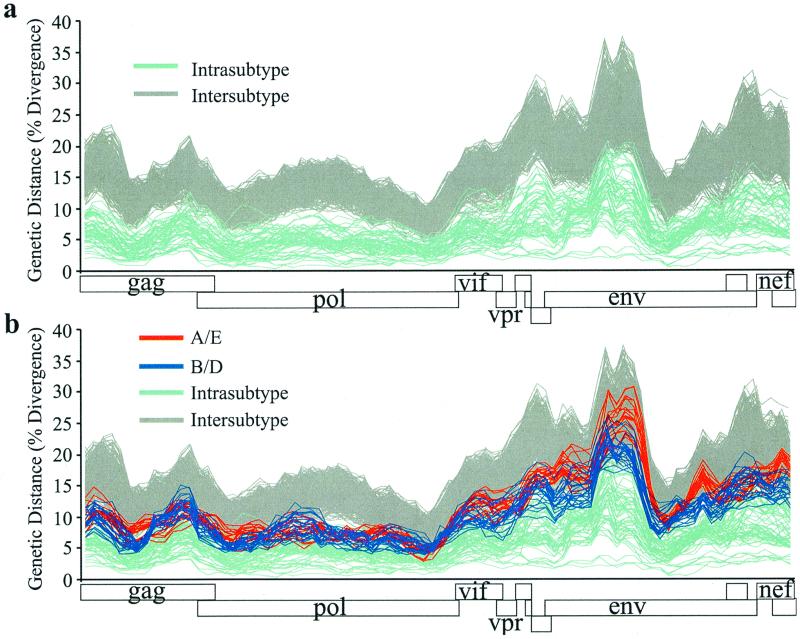
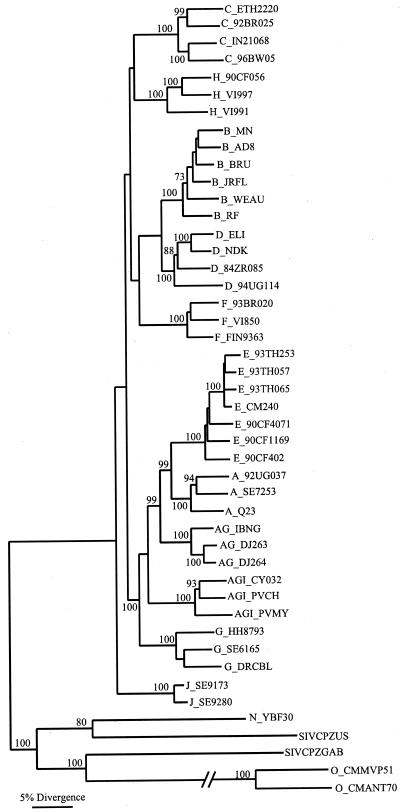
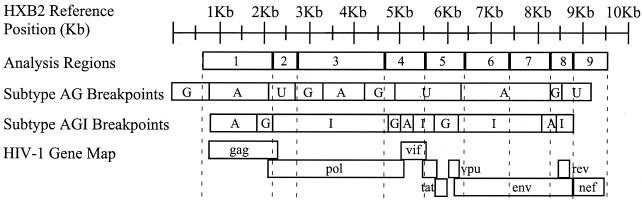
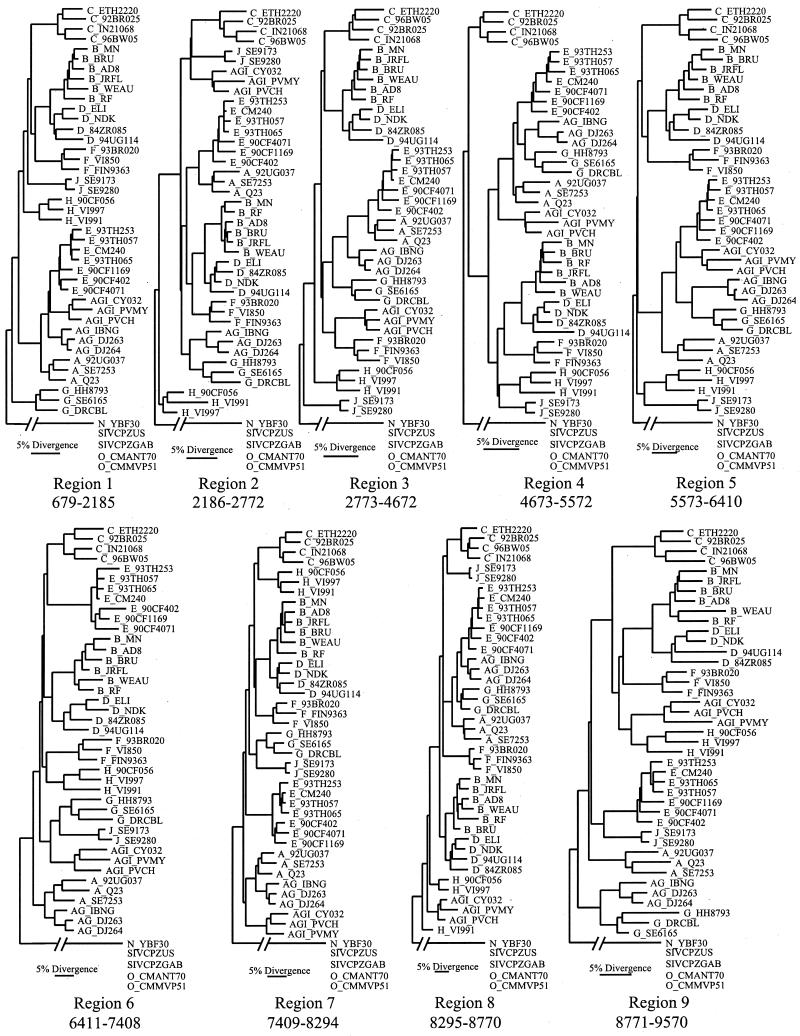
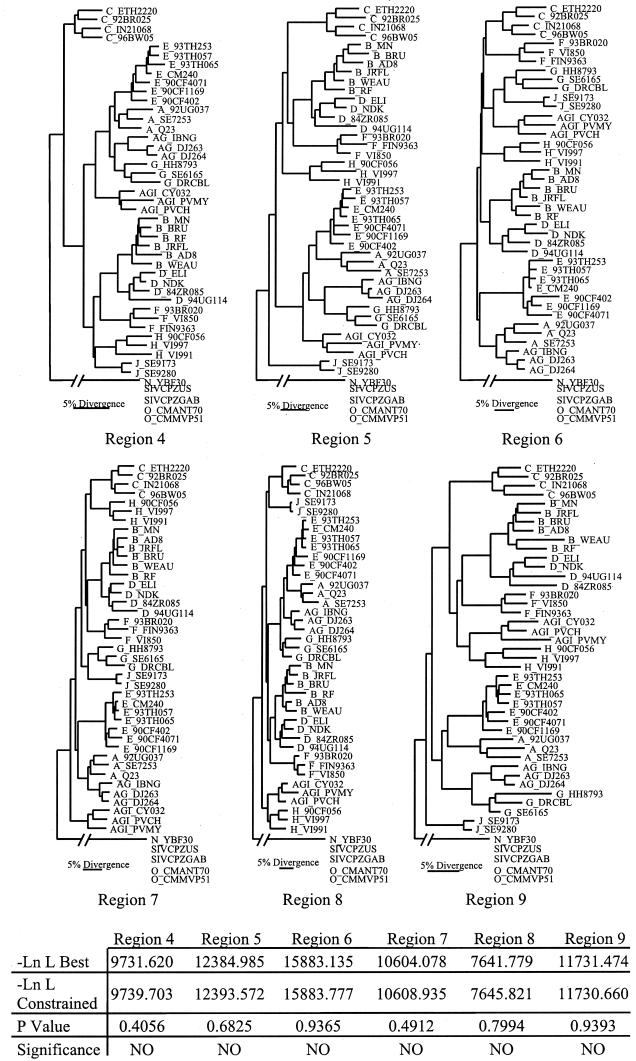
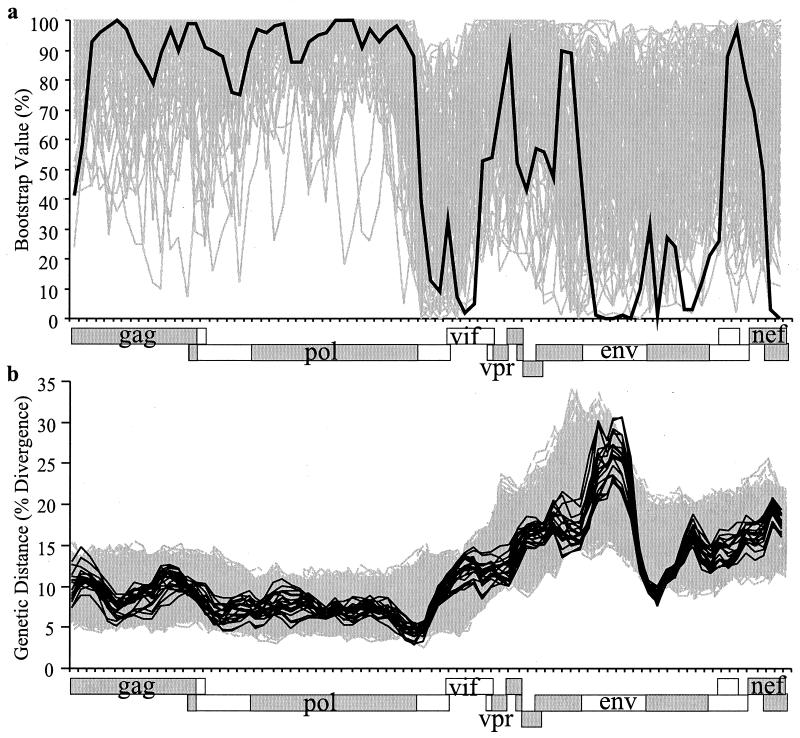
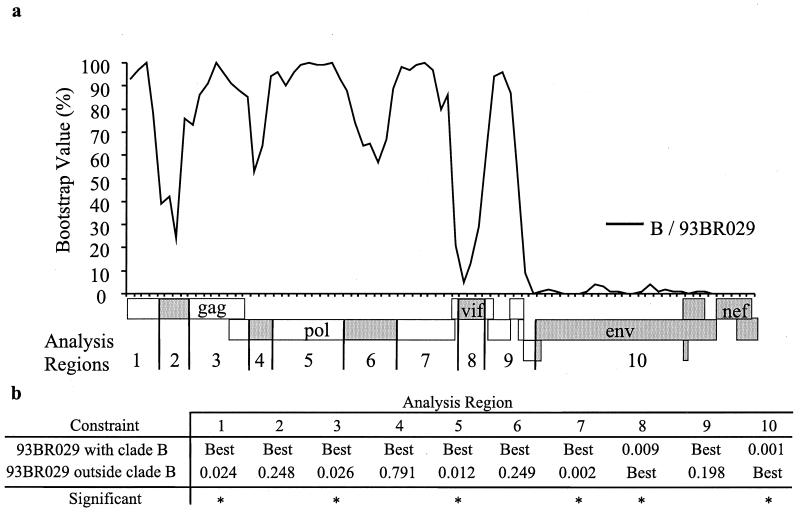
The human immunodeficiency virus type 1 (HIV-1) epidemic in Southeast Asia has been largely due to the emergence of clade E (HIV-1E). It has been suggested that HIV-1E is derived from a recombinant lineage of subtype A (HIV-1A) and subtype E, with multiple breakpoints along the E genome. We obtained complete genome sequences of clade E viruses from Thailand (93TH057 and 93TH065) and from the Central African Republic (90CF11697 and 90CF4071), increasing the total number of HIV-1E complete genome sequences available to seven. Phylogenetic analysis of complete genomes showed that subtypes A and E are themselves monophyletic, although together they also form a larger monophyletic group. The apparent phylogenetic incongruence at different regions of the genome that was previously taken as evidence of recombination is shown to be not statistically significant. Furthermore, simulations indicate that bootscanning and pairwise distance results, previously used as evidence for recombination, can be misleading, particularly when there are differences in substitution or evolutionary rates across the genomes of different subtypes. Taken jointly, our analyses suggest that there is inadequate support for the hypothesis that subtype E variants are derived from a recombinant lineage. In contrast, many other HIV strains claimed to have a recombinant origin, including viruses for which only a single parental strain was employed for analysis, do indeed satisfy the statistical criteria we propose. Thus, while intersubtype recombinant HIV strains are indeed circulating, the criteria for assigning a recombinant origin to viral structures should include statistical testing of alternative hypotheses to avoid inappropriate assignments that would obscure the true evolutionary properties of these viruses.
Figures

References
-
- Carr J K, Foley B T, Leitner T, Salminen M, Korber B, McCutchan F. Reference sequences representing the principal genetic diversity of HIV-1 in the pandemic. Vol. 1999. Los Alamos, N.M: Los Alamos National Laboratory; 1999.
-
- Carr J K, Salminen M O, Albert J, Sanders-Buell E, Gotte D, Birx D L, McCutchan F E. Full genome sequences of human immunodeficiency virus type 1 subtypes G and A/G intersubtype recombinants. Virology. 1998;247:22–31. - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
