

Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members
- PMID: 11050115
- PMCID: PMC6772747
- DOI: 10.1523/JNEUROSCI.20-21-07951.2000
Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members
Abstract
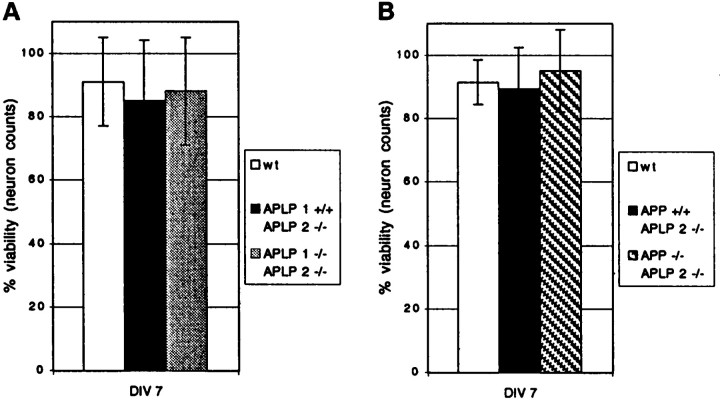
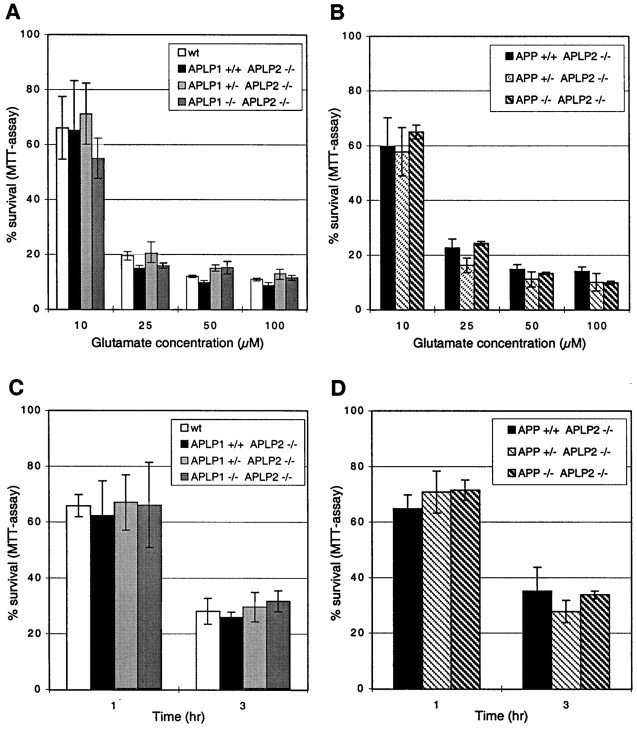
The amyloid precursor protein (APP) involved in Alzheimer's disease is a member of a larger gene family including amyloid precursor-like proteins APLP1 and APLP2. We generated and examined the phenotypes of mice lacking individual or all possible combinations of APP family members to assess potential functional redundancies within the gene family. Mice deficient for the nervous system-specific APLP1 protein showed a postnatal growth deficit as the only obvious abnormality. In contrast to this minor phenotype, APLP2(-/-)/APLP1(-/-) and APLP2(-/-)/APP(-/-) mice proved lethal early postnatally. Surprisingly, APLP1(-/-)/APP(-/-) mice were viable, apparently normal, and showed no compensatory upregulation of APLP2 expression. These data indicate redundancy between APLP2 and both other family members and corroborate a key physiological role for APLP2. This view gains further support by the observation that APLP1(-/-)/APP(-/-)/APLP2(+/-) mice display postnatal lethality. In addition, they provide genetic evidence for at least some distinct physiological roles of APP and APLP2 by demonstrating that combinations of single knock-outs with the APLP1 mutation resulted in double mutants of clearly different phenotypes, being either lethal, or viable. None of the lethal double mutants displayed, however, obvious histopathological abnormalities in the brain or any other organ examined. Moreover, cortical neurons from single or combined mutant mice showed unaltered survival rates under basal culture conditions and unaltered susceptibility to glutamate excitotoxicity in vitro.
Figures
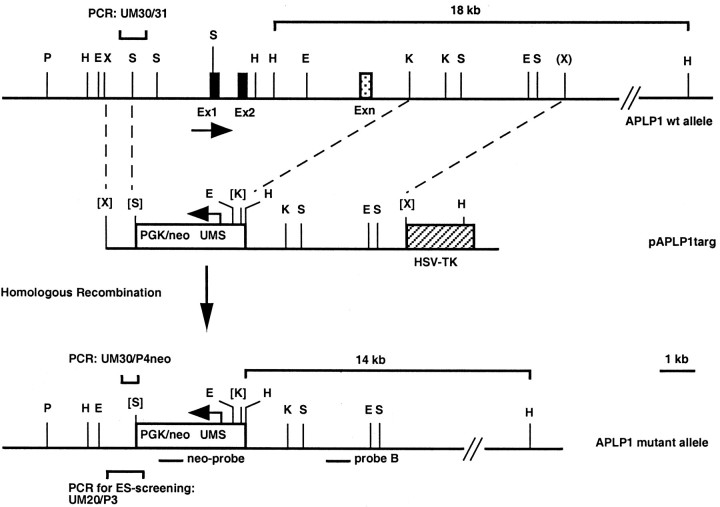
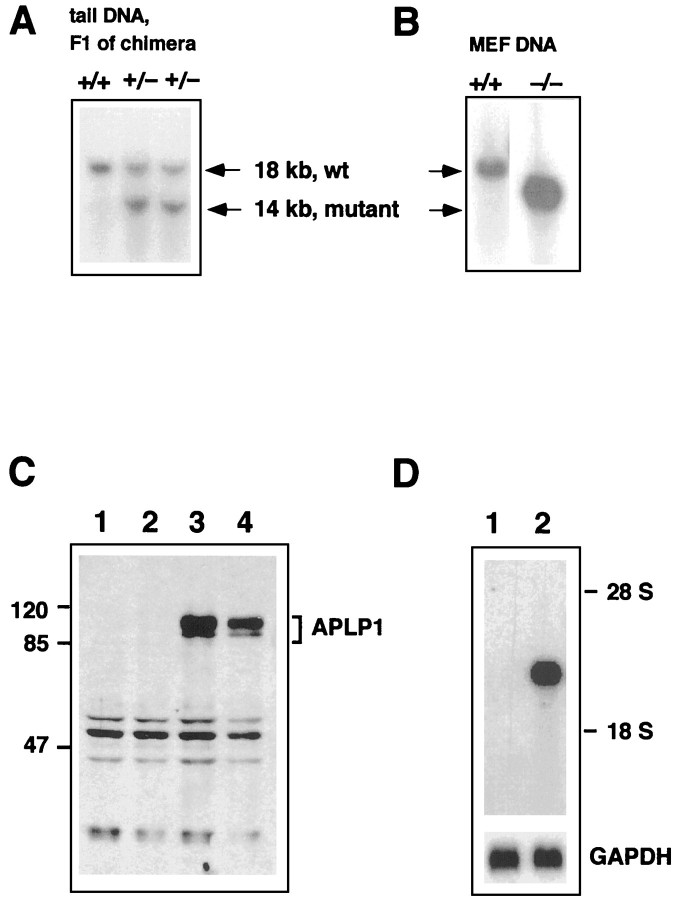
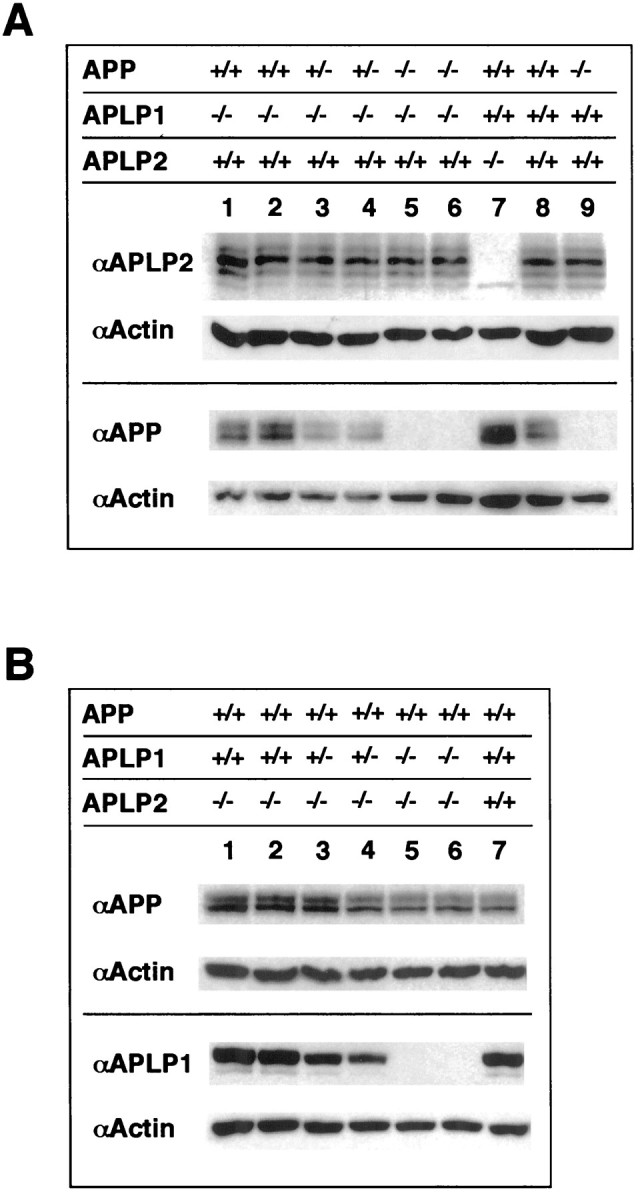
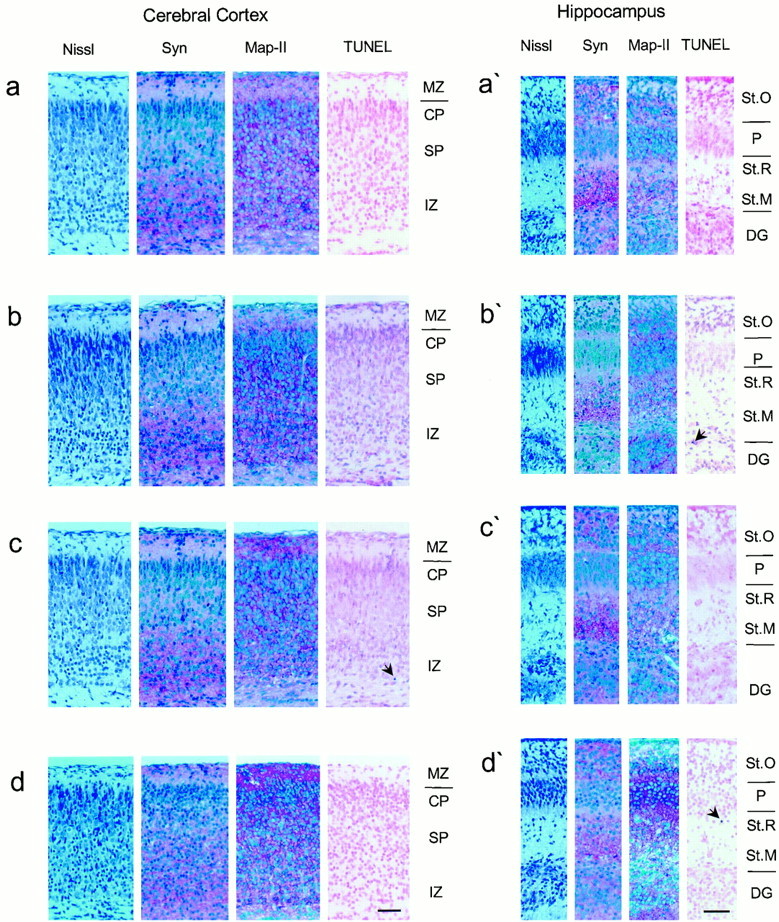

References
-
- Banker G, Goslin K. Culturing nerve cells. MIT; Cambridge, MA: 1998.
-
- Fox WM. Reflex-ontogeny and behavioural development of the mouse. Anim Behav. 1965;13:234–241. - PubMed
-
- Furukawa K, Mattson MP. Secreted amyloid precursor protein alpha selectively suppresses N-methyl-d-aspartate currents in hippocampal neurons: involvement of cyclic GMP. Neuroscience. 1998;83:429–438. - PubMed
-
- Furukawa K, Barger SW, Blalock EM, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted beta-amyloid-precursor protein. Nature. 1996;379:74–78. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous