

Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis
- PMID: 11050163
- PMCID: PMC18805
- DOI: 10.1073/pnas.220417997
Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis
Abstract
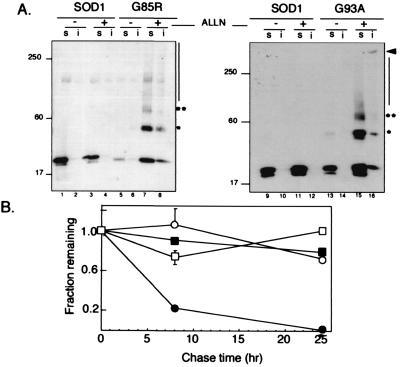
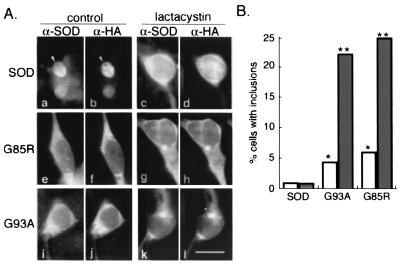
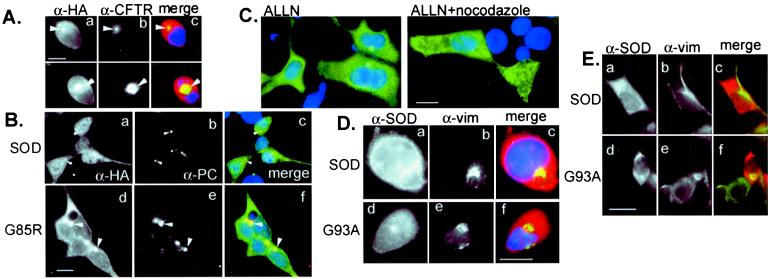
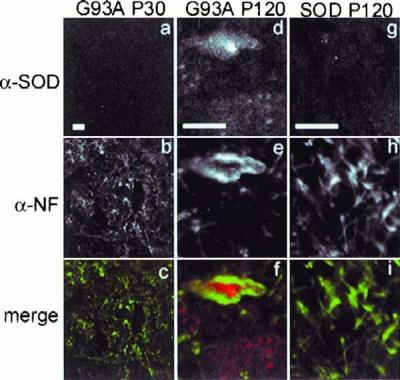
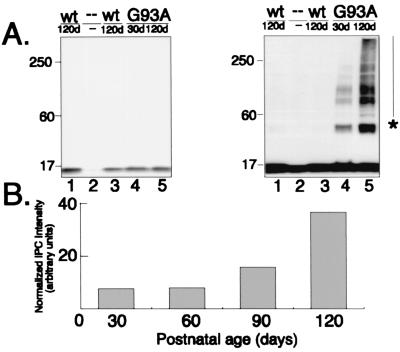
Deposition of aggregated protein into neurofilament-rich cytoplasmic inclusion bodies is a common cytopathological feature of neurodegenerative disease. How-or indeed whether-protein aggregation and inclusion body formation cause neurotoxicity are presently unknown. Here, we show that the capacity of superoxide dismutase (SOD) to aggregate into biochemically distinct, high molecular weight, insoluble protein complexes (IPCs) is a gain of function associated with mutations linked to autosomal dominant familial amyotrophic lateral sclerosis. SOD IPCs are detectable in spinal cord extracts from transgenic mice expressing mutant SOD several months before inclusion bodies and motor neuron pathology are apparent. Sequestration of mutant SOD into cytoplasmic inclusion bodies resembling aggresomes requires retrograde transport on microtubules. These data indicate that aggregation and inclusion body formation are mechanistically and temporally distinct processes.
Figures
References
-
- Siddique T. Adv Neurol. 1991;56:227–231. - PubMed
-
- Emery A, Holloway S. In: Human Motor Neuron Diseases. Rowland L, editor. New York: Raven; 1982. pp. 139–147.
-
- Rosen D R, Siddique T, Patterson D, Figlewicz D A, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan J P, Deng H X, et al. Nature (London) 1993;362:59–62. - PubMed
-
- Siddique T, Nijhawan D, Hentati A. Neurology. 1996;47:S27–S35. - PubMed
-
- Reaume A G, Elliott J L, Hoffman E K, Kowall N W, Ferrante R J, Siwek D F, Wilcox H M, Flood D G, Beal M F, Brown R H, Jr, et al. Nat Genet. 1996;13:43–47. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
