

Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion
- PMID: 11055897
- PMCID: PMC1287919
- DOI: 10.1086/316911
Active intestinal chloride secretion in human carriers of cystic fibrosis mutations: an evaluation of the hypothesis that heterozygotes have subnormal active intestinal chloride secretion
Abstract
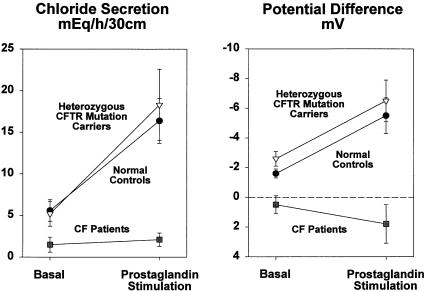
To explain the very high frequency of cystic fibrosis (CF) mutations in most populations of European descent, it has been proposed that CF heterozygotes have a survival advantage when infected with Vibrio cholerae or Escherichia coli, the toxins of which induce diarrhea by stimulation of active intestinal chloride secretion. Two assumptions underlie this hypothesis: (1) chloride conductance by the CF transmembrane conductance regulator (CFTR) is the rate-limiting step for active intestinal chloride secretion at all levels of expression, from approximately zero in patients with CF to normal levels in people who are not carriers of a mutation; and (2) heterozygotes have smaller amounts of functional intestinal CFTR than do people who are not carriers, and heterozygotes therefore secrete less chloride when exposed to secretagogues. The authors used an intestinal perfusion technique to measure in vivo basal and prostaglandin-stimulated jejunal chloride secretion in normal subjects, CF heterozygotes, and patients with CF. Patients with CF had essentially no active chloride secretion in the basal state, and secretion was not stimulated by a prostaglandin analogue. However, CF heterozygotes secreted chloride at the same rate as did people without a CF mutation. If heterozygotes are assumed to have less-than-normal intestinal CFTR function, these results mean that CFTR expression is not rate limiting for active chloride secretion in heterozygotes. The results do not support the theory that the very high frequency of CF mutations is due to a survival advantage that is conferred on heterozygotes who contract diarrheal illnesses mediated by intestinal hypersecretion of chloride.
Figures
Similar articles
-
Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model.Science. 1994 Oct 7;266(5182):107-9. doi: 10.1126/science.7524148. Science. 1994. PMID: 7524148
-
Chloride transport in nasal ciliated cells of cystic fibrosis heterozygotes.Am J Respir Crit Care Med. 2005 May 1;171(9):1026-31. doi: 10.1164/rccm.200406-740OC. Epub 2005 Feb 11. Am J Respir Crit Care Med. 2005. PMID: 15709055
-
Residual chloride secretion in intestinal tissue of deltaF508 homozygous twins and siblings with cystic fibrosis. The European CF Twin and Sibling Study Consortium.Gastroenterology. 2000 Jul;119(1):32-40. doi: 10.1053/gast.2000.8524. Gastroenterology. 2000. PMID: 10889152
-
[New therapeutic developments in cystic fibrosis].Arch Pediatr. 2016 Dec;23(12S):12S47-12S53. doi: 10.1016/S0929-693X(17)30062-3. Arch Pediatr. 2016. PMID: 28231894 Review. French.
-
CFTR chloride channel drug discovery--inhibitors as antidiarrheals and activators for therapy of cystic fibrosis.Curr Pharm Des. 2006;12(18):2235-47. doi: 10.2174/138161206777585148. Curr Pharm Des. 2006. PMID: 16787252 Review.
Cited by
-
Origin and spread of the 1278insTATC mutation causing Tay-Sachs disease in Ashkenazi Jews: genetic drift as a robust and parsimonious hypothesis.Hum Genet. 2004 Mar;114(4):366-76. doi: 10.1007/s00439-003-1072-8. Epub 2004 Jan 15. Hum Genet. 2004. PMID: 14727180
-
Evaluating candidate agents of selective pressure for cystic fibrosis.J R Soc Interface. 2007 Feb 22;4(12):91-8. doi: 10.1098/rsif.2006.0154. J R Soc Interface. 2007. PMID: 17015291 Free PMC article.
-
Common CFTR gene variants influence body composition and survival in rural Ghana.Hum Genet. 2010 Feb;127(2):201-6. doi: 10.1007/s00439-009-0762-2. Epub 2009 Nov 5. Hum Genet. 2010. PMID: 19890664 Free PMC article.
-
Selective inhibition of intestinal guanosine 3',5'-cyclic monophosphate signaling by small-molecule protein kinase inhibitors.J Biol Chem. 2018 May 25;293(21):8173-8181. doi: 10.1074/jbc.RA118.002835. Epub 2018 Apr 13. J Biol Chem. 2018. PMID: 29653944 Free PMC article.
-
Intestinal current measurement versus nasal potential difference measurements for diagnosis of cystic fibrosis: a case-control study.BMC Pulm Med. 2014 Oct 4;14:156. doi: 10.1186/1471-2466-14-156. BMC Pulm Med. 2014. PMID: 25280757 Free PMC article.
References
Electronic-Database Information
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.gov/omim (for CF [MIM 219700] and CFTR [MIM 602421])
-
- Genome Database, http://gdbwww.gdb.org (for CFTR [accession number 120584])
References
-
- Baxter PS, Goldhill J, Hardcastle J, Hardcastle PT, Taylor CJ (1988) Accounting for cystic fibrosis. Nature 335:211 - PubMed
-
- Baxter PS, Wilson AJ, Read NW, Hardcastle J, Hardcastle PT, Taylor CJ (1989) Abnormal jejunal potential difference in cystic fibrosis. Lancet 333:464–466 - PubMed
-
- Berschneider HM, Knowles MR, Azizkhan RG, Boucher RC, Tobey NA, Orlando RC, Powell DW (1988) Altered intestinal chloride transport in cystic fibrosis. FASEB J 2:2625–2629 - PubMed
-
- Bertranpetit J, Calafell F (1996) Genetic and geographical variability in cystic fibrosis: evolutionary considerations. Variation in the human genome. (Ciba Foundation Symposium 197). John Wiley, Chichester, United Kingdom, pp 97–118 - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
