

Functional consequences of Rett syndrome mutations on human MeCP2
- PMID: 11058114
- PMCID: PMC113135
- DOI: 10.1093/nar/28.21.4172
Functional consequences of Rett syndrome mutations on human MeCP2
Abstract
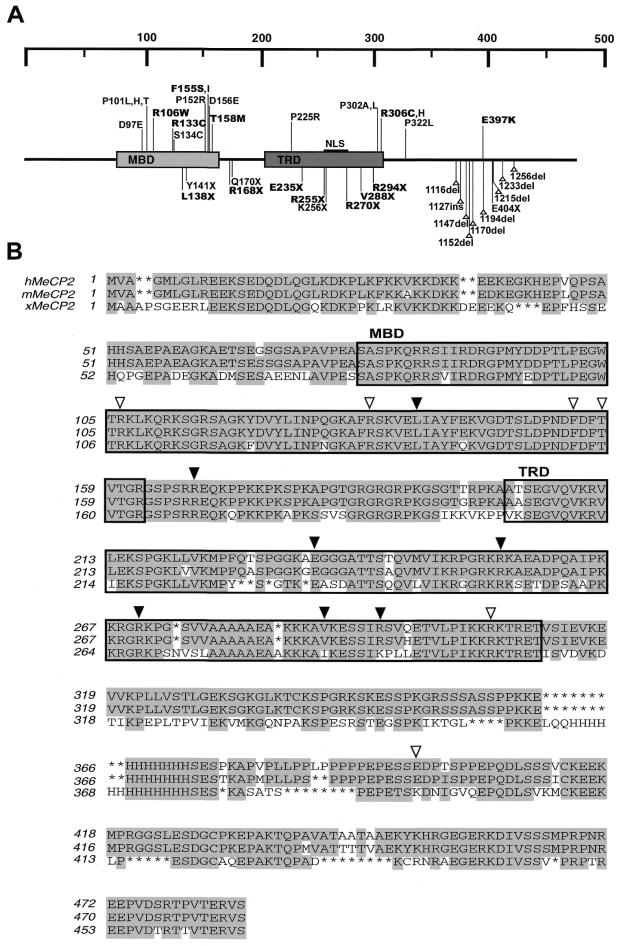
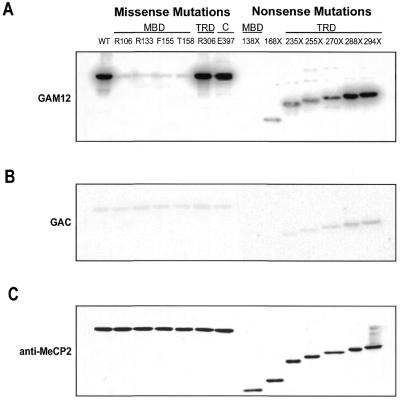
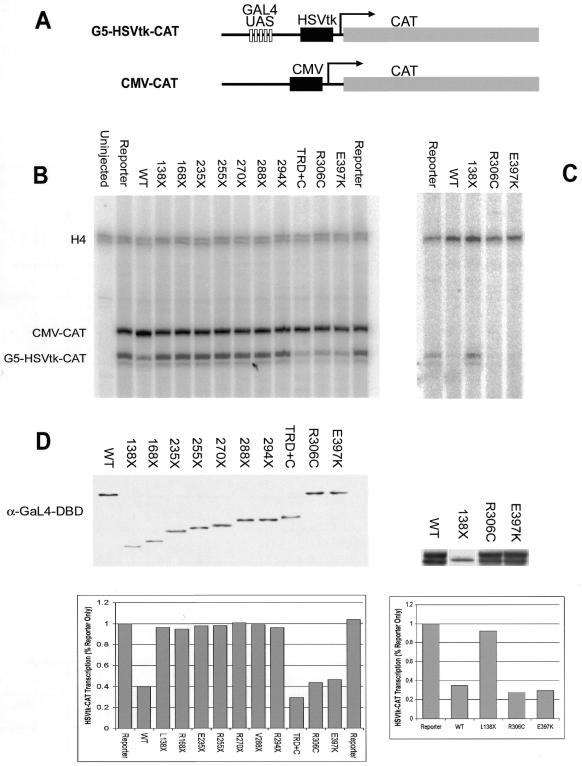
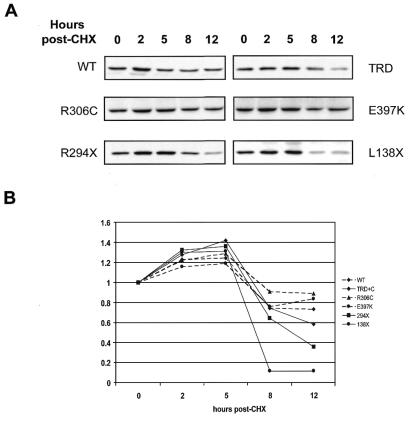
The neurodevelopmental disorder known as Rett syndrome has recently been linked to the methyl-CpG-binding transcriptional repressor, MeCP2. In this report we examine the consequences of these mutations on the function of MeCP2. The ability to bind specifically to methylated DNA and the transcription repression capabilities are tested, as well as the stability of proteins in vivo. We find that all missense mutations (R106W, R133C, F155S, T158M) within the methyl-binding domain impair selectivity for methylated DNA, and that all nonsense mutations (L138X, R168X, E235X, R255X, R270X, V288X, R294X) that truncate all or some of the transcriptional repression domain (TRD) affect the ability to repress transcription and have decreased levels of stability in vivo. Two missense mutations, one in the TRD (R306C) and one in the C-terminus (E397K), had no noticeable effects on MeCP2 function. Together, these results provide evidence of how Rett syndrome mutations can affect distinct functions of MeCP2 and give insight into these mutations that may contribute to the disease.
Figures
Similar articles
-
Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA.Biochemistry. 2000 Jun 20;39(24):7100-6. doi: 10.1021/bi0001271. Biochemistry. 2000. PMID: 10852707
-
A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome.J Mol Med (Berl). 2004 Feb;82(2):135-43. doi: 10.1007/s00109-003-0497-9. Epub 2003 Nov 15. J Mol Med (Berl). 2004. PMID: 14618241
-
Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots.Am J Hum Genet. 1999 Dec;65(6):1520-9. doi: 10.1086/302690. Am J Hum Genet. 1999. PMID: 10577905 Free PMC article.
-
Rett syndrome: a surprising result of mutation in MECP2.Hum Mol Genet. 2000 Oct;9(16):2365-75. doi: 10.1093/hmg/9.16.2365. Hum Mol Genet. 2000. PMID: 11005791 Review.
-
The biological functions of the methyl-CpG-binding protein MeCP2 and its implication in Rett syndrome.Brain Dev. 2001 Dec;23 Suppl 1:S32-7. doi: 10.1016/s0387-7604(01)00333-3. Brain Dev. 2001. PMID: 11738839 Review.
Cited by
-
Binding Analysis of Methyl-CpG Binding Domain of MeCP2 and Rett Syndrome Mutations.ACS Chem Biol. 2016 Oct 21;11(10):2706-2715. doi: 10.1021/acschembio.6b00450. Epub 2016 Aug 8. ACS Chem Biol. 2016. PMID: 27356039 Free PMC article.
-
Methyl-CpG-binding (SmMBD2/3) and chromobox (SmCBX) proteins are required for neoblast proliferation and oviposition in the parasitic blood fluke Schistosoma mansoni.PLoS Pathog. 2018 Jun 28;14(6):e1007107. doi: 10.1371/journal.ppat.1007107. eCollection 2018 Jun. PLoS Pathog. 2018. PMID: 29953544 Free PMC article.
-
Rett syndrome and the impact of MeCP2 associated transcriptional mechanisms on neurotransmission.Biol Psychiatry. 2009 Feb 1;65(3):204-10. doi: 10.1016/j.biopsych.2008.10.036. Epub 2008 Dec 5. Biol Psychiatry. 2009. PMID: 19058783 Free PMC article. Review.
-
Generation and characterization of rat and mouse monoclonal antibodies specific for MeCP2 and their use in X-inactivation studies.PLoS One. 2011;6(11):e26499. doi: 10.1371/journal.pone.0026499. Epub 2011 Nov 28. PLoS One. 2011. PMID: 22140431 Free PMC article.
-
Biochemical analysis of histone deacetylase-independent transcriptional repression by MeCP2.J Biol Chem. 2013 Mar 8;288(10):7096-104. doi: 10.1074/jbc.M112.438697. Epub 2013 Jan 24. J Biol Chem. 2013. PMID: 23349465 Free PMC article.
References
-
- Amir R.E., Van den Veyver,I.B., Wan,M., Tran,C.Q., Francke,U. and Zoghbi,H.Y. (1999) Nature Genet., 23, 185–188. - PubMed
-
- Cheadle J.P., Gill,H., Fleming,N., Maynard,J., Kerr,A., Leonard,H., Krawczak,M., Cooper,D.N., Lynch,S., Thomas,N., Hughes,H., Hulten,M., Ravine,D., Sampson,J.R. and Clarke,A. (2000) Hum. Mol. Genet., 9, 1119–1129. - PubMed
-
- Huppke P., Laccone,F., Kramer,N., Engel,W. and Hanefeld,F. (2000) Hum. Mol. Genet., 9, 1369–1375. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
