

Crystal structure of the human alpha-thrombin-haemadin complex: an exosite II-binding inhibitor
- PMID: 11060016
- PMCID: PMC305786
- DOI: 10.1093/emboj/19.21.5650
Crystal structure of the human alpha-thrombin-haemadin complex: an exosite II-binding inhibitor
Abstract
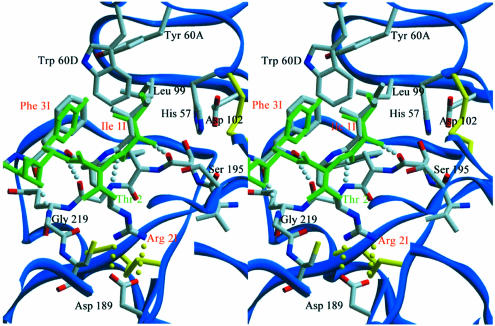
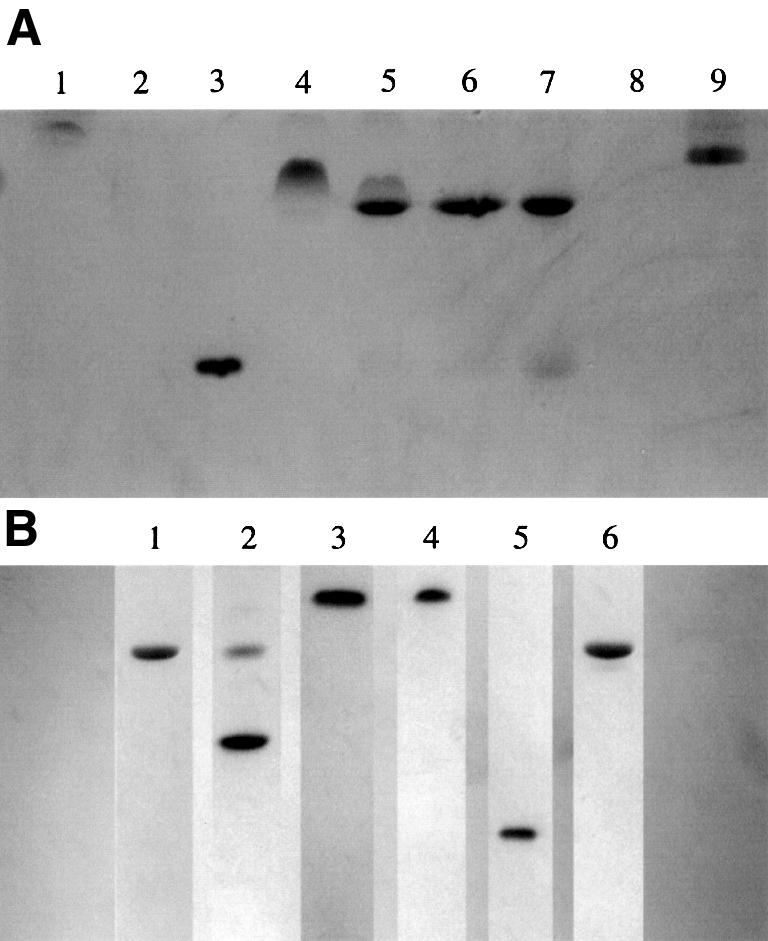
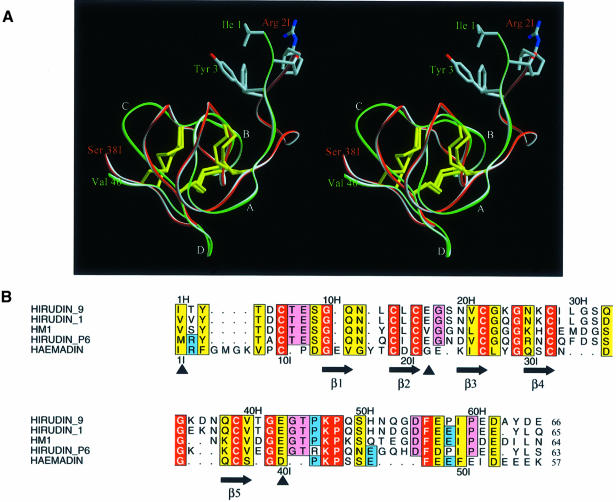
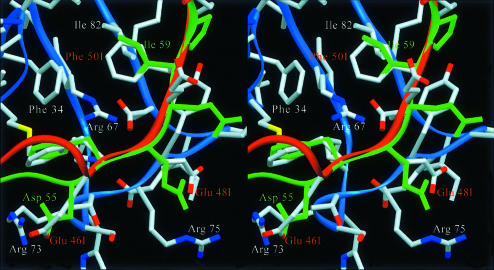
The serine proteinase alpha-thrombin plays a pivotal role in the regulation of blood fluidity, and therefore constitutes a primary target in the treatment of various haemostatic disorders. Haemadin is a slow tight- binding thrombin inhibitor from the land-living leech Haemadipsa sylvestris. Here we present the 3.1 A crystal structure of the human alpha-thrombin- haemadin complex. The N-terminal segment of haemadin binds to the active site of thrombin, forming a parallel beta-strand with residues Ser214-Gly216 of the proteinase. This mode of binding is similar to that observed in another leech-derived inhibitor, hirudin. In contrast to hirudin, however, the markedly acidic C-terminal peptide of haemadin does not bind the fibrinogen-recognition exosite, but interacts with the heparin-binding exosite of thrombin. Thus, haemadin binds to thrombin according to a novel mechanism, despite an overall structural similarity with hirudin. Haemadin inhibits both free and thrombomodulin-bound alpha-thrombin, but not intermediate activation forms such as meizothrombin. This specific anticoagulant ability of haemadin makes it an ideal candidate for an antithrombotic agent, as well as a starting point for the design of novel antithrombotics.
Figures
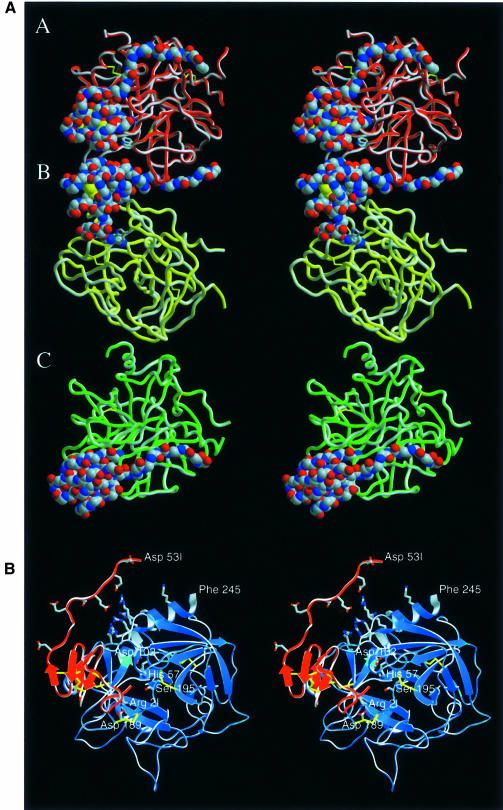
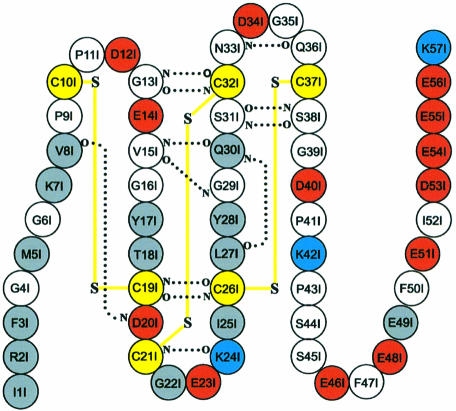
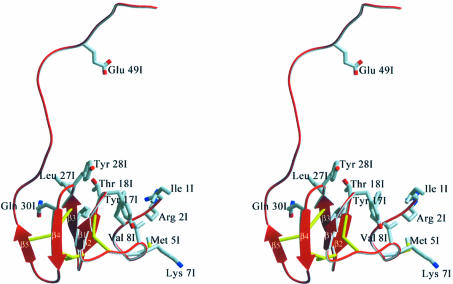
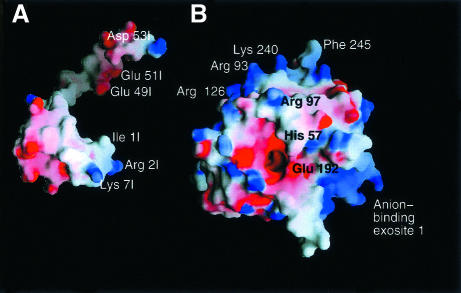
References
-
- Arocas V., Zingali,R.B., Guillin,M.-C., Bon,C. and Jandrot-Perrus,M. (1996) Bothrojaracin: a potent two-site-directed thrombin inhibitor. Biochemistry, 35, 9083–9089. - PubMed
-
- Barton G.J. (1993) ALSCRIPT: a tool to format multiple sequence alignments. Protein Eng., 6, 37–40. - PubMed
-
- Betz A., Hofsteenge,J. and Stone,S.R. (1992) Interaction of the N-terminal region of hirudin with the active-site cleft of thrombin. Biochemistry, 31, 4557–4562. - PubMed
-
- Bode C., Nordt,T.K. and Runge,M.S. (1994) Thrombolytic therapy in acute myocardial infarction—selected recent developments. Ann. Hematol., 69, S35–40. - PubMed
-
- Bode W. and Huber,R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem., 204, 433–451. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
