

Is "junk" DNA mostly intron DNA?
- PMID: 11076852
- PMCID: PMC310976
- DOI: 10.1101/gr.148900
Is "junk" DNA mostly intron DNA?
Abstract
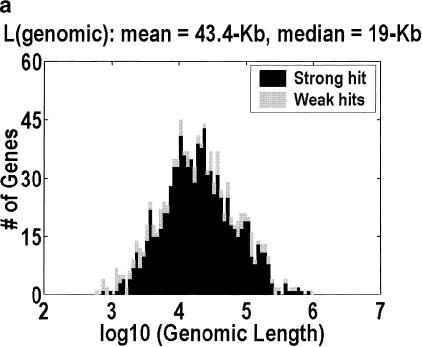
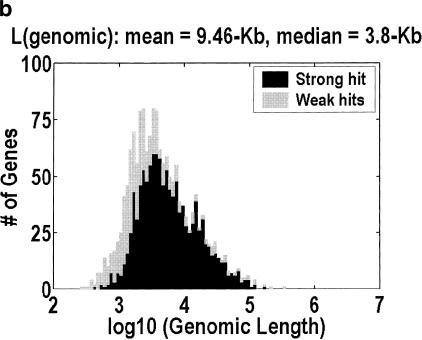
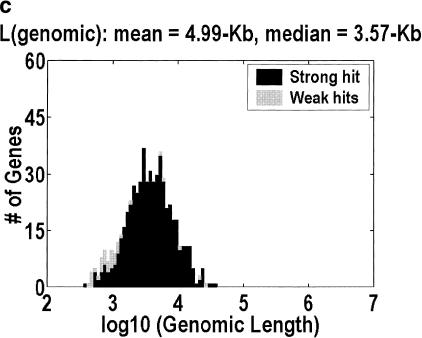
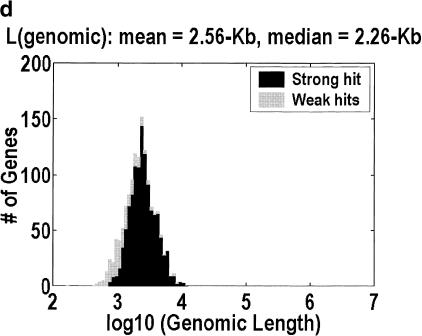
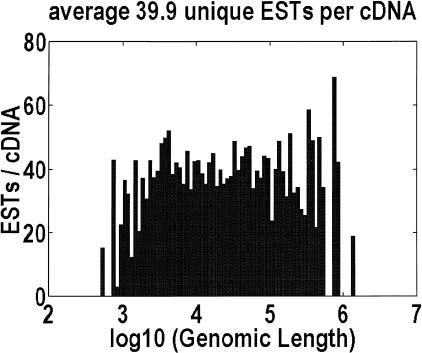
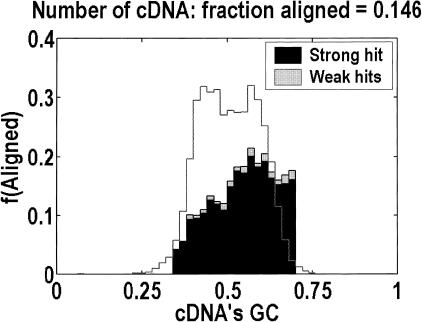
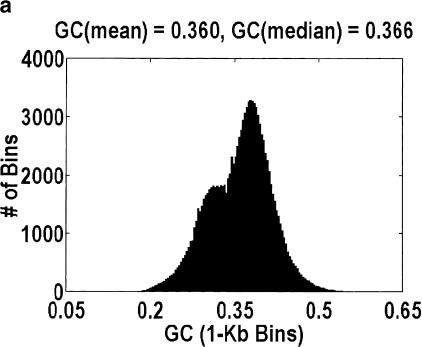
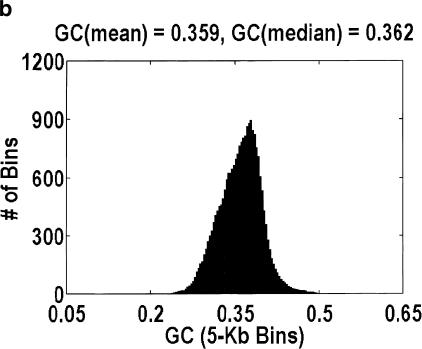
Among higher eukaryotes, very little of the genome codes for protein. What is in the rest of the genome, or the "junk" DNA, that, in Homo sapiens, is estimated to be almost 97% of the genome? Is it possible that much of this "junk" is intron DNA? This is not a question that can be answered just by looking at the published data, even from the finished genomes. One cannot assume that there are no genes in a sequenced region, just because no genes were annotated. We introduce another approach to this problem, based on an analysis of the cDNA-to-genomic alignments, in all of the complete or nearly-complete genomes from the multicellular organisms. Our conclusion is that, in animals but not in plants, most of the "junk" is intron DNA.
Figures
References
-
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. - PubMed
-
- Burset M, Guigo R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–367. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases