

Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome
- PMID: 11078474
- PMCID: PMC1287915
- DOI: 10.1086/316915
Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome
Abstract
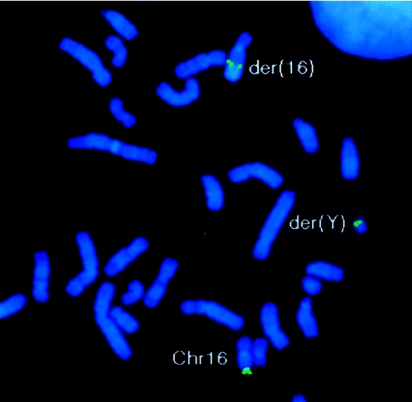
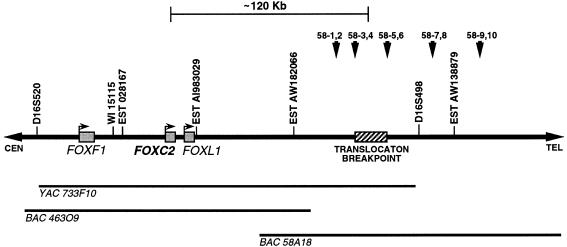
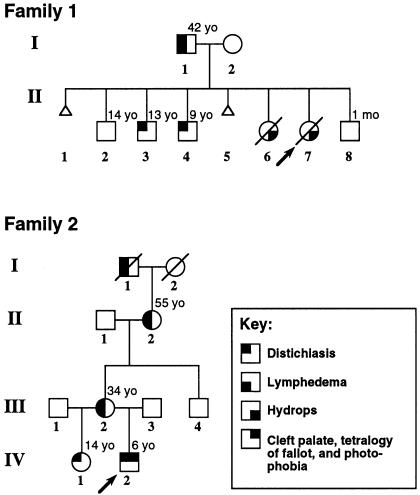
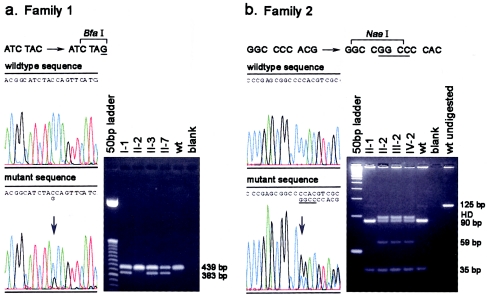
Lymphedema-distichiasis (LD) is an autosomal dominant disorder that classically presents as lymphedema of the limbs, with variable age at onset, and double rows of eyelashes (distichiasis). Other complications may include cardiac defects, cleft palate, extradural cysts, and photophobia, suggesting a defect in a gene with pleiotrophic effects acting during development. We previously reported neonatal lymphedema, similar to that in Turner syndrome, associated with a t(Y;16)(q12;q24.3) translocation. A candidate gene was not found on the Y chromosome, and we directed our efforts toward the chromosome 16 breakpoint. Subsequently, a gene for LD was mapped, by linkage studies, to a 16-cM region at 16q24.3. By FISH, we determined that the translocation breakpoint was within this critical region and further narrowed the breakpoint to a 20-kb interval. Because the translocation did not appear to interrupt a gene, we considered candidate genes in the immediate region that might be inactivated by position effect. In two additional unrelated families with LD, we identified inactivating mutations-a nonsense mutation and a frameshift mutation-in the FOXC2 (MFH-1) gene. FOXC2 is a member of the forkhead/winged-helix family of transcription factors, whose members are involved in diverse developmental pathways. FOXC2 knockout mice display cardiovascular, craniofacial, and vertebral abnormalities similar to those seen in LD syndrome. Our findings show that FOXC2 haploinsufficiency results in LD. FOXC2 represents the second known gene to result in hereditary lymphedema, and LD is only the second hereditary disorder known to be caused by a mutation in a forkhead-family gene.
Figures
References
Electronic-Database Information
-
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for FOXC2 [accession number NM_005251] and FOXL1 [accession number AF315075])
-
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for Milroy disease [MIM 153100] and LD [MIM 153400])
References
-
- Baskaran N, Kandpal RP, Bhargava AK, Glynn MW, Bale A, Weissman SM (1996) Uniform amplification of a mixture of deoxyribonucleic acids with varying GC content. Genome Res 6:633–638 - PubMed
-
- Corbett CRR, Dale RF, Coltart DJ, Kinmonth JB (1982) Congenital heart disease in patients with primary lymphoedemas. Lymphology 15:85–90 - PubMed
-
- Dagenais SL, Guevara-Fujita M, Loechel R, Burgess AC, Miller DE, Yuzbasiyan-Gurkan V, Brewer GJ, Glover TW (1999) The canine copper toxicosis locus is not syntenic with ATP7B or ATX1 and maps to a region showing homology to human 2p21. Mamm Genome 10:753–756 - PubMed
-
- Erickson RP, Hudgins L, Stone JF, Schmidt S, Wilke C, Glover TW (1995) A “balanced” Y;16 translocation associated with Turner-like neonatal lymphedema suggests the location of a potential anti-Turner gene on the Y chromosome. Cytogenet Cell Genet 71:163–167 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
