

Somatic hypermutation in the absence of DNA-dependent protein kinase catalytic subunit (DNA-PK(cs)) or recombination-activating gene (RAG)1 activity
- PMID: 11085752
- PMCID: PMC2193187
- DOI: 10.1084/jem.192.10.1509
Somatic hypermutation in the absence of DNA-dependent protein kinase catalytic subunit (DNA-PK(cs)) or recombination-activating gene (RAG)1 activity
Abstract
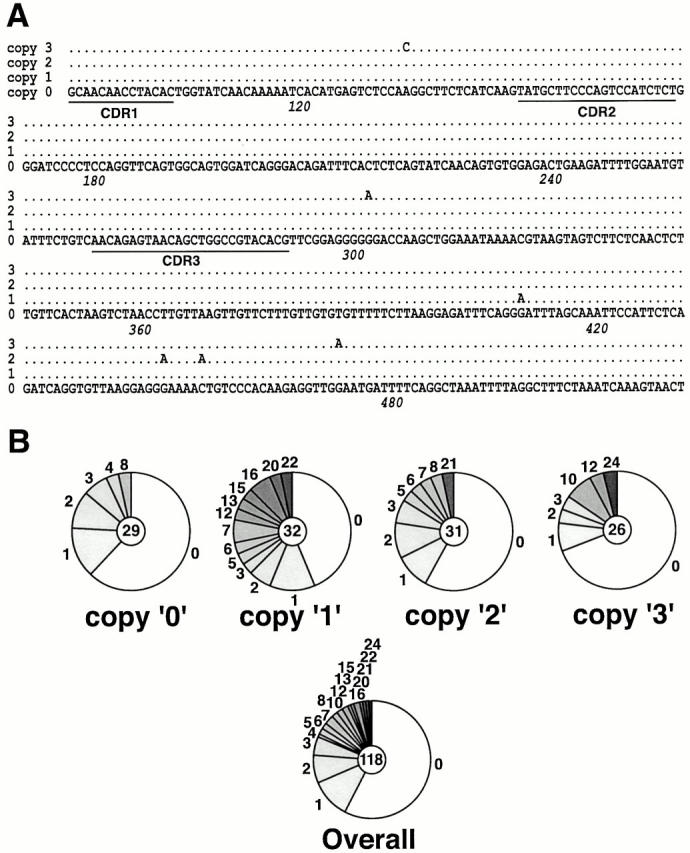
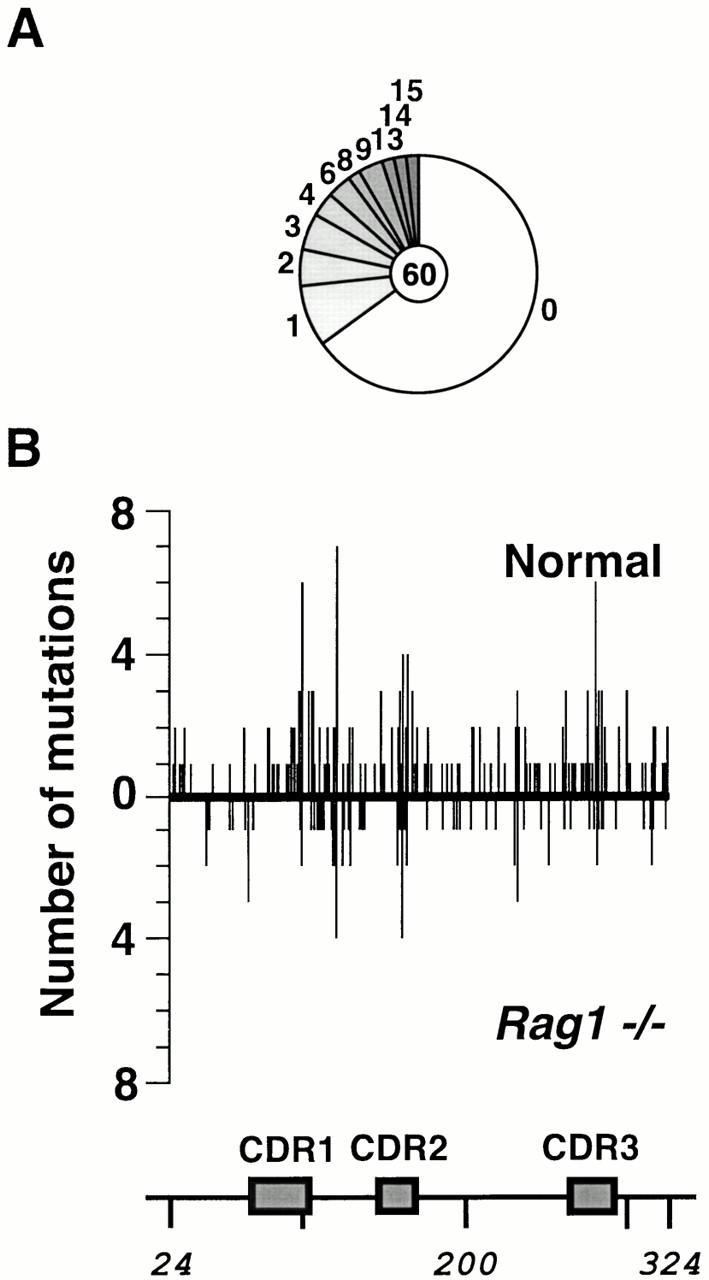
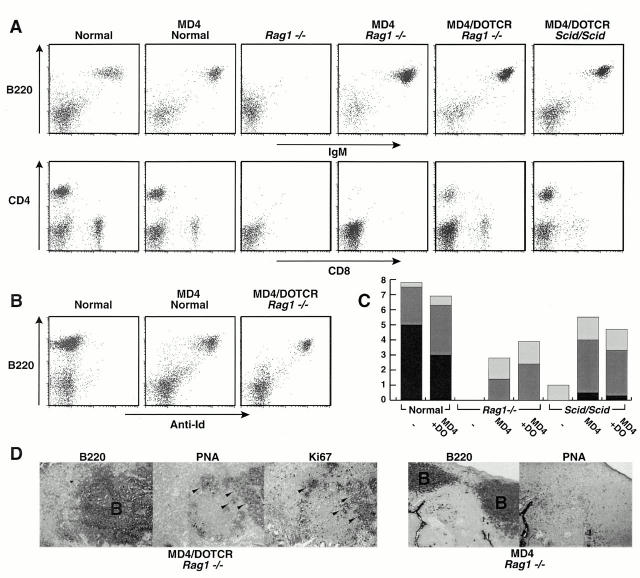
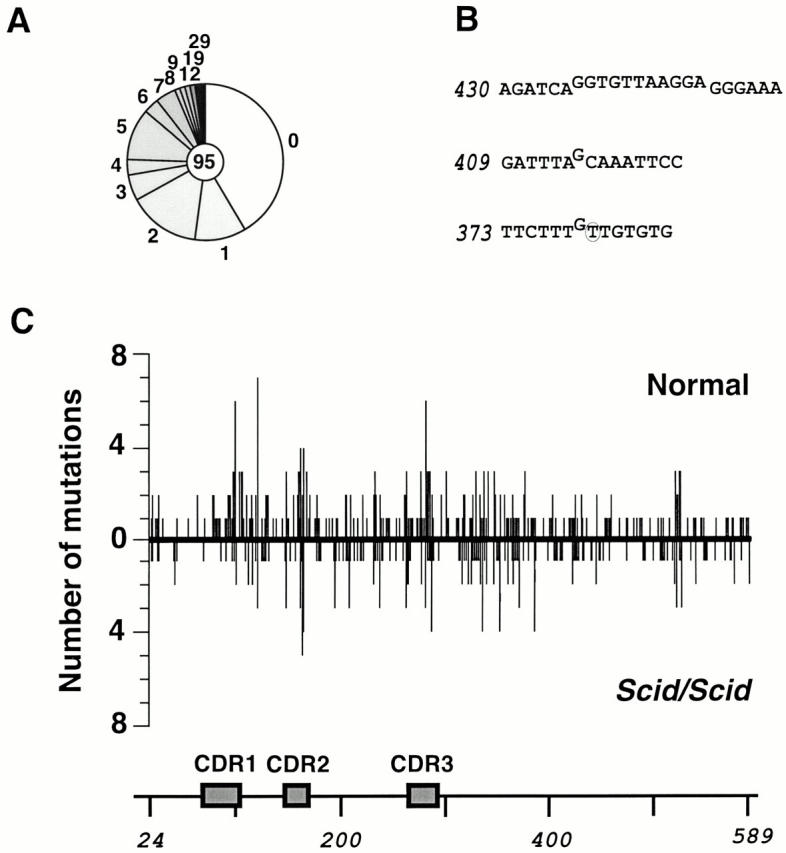
Somatic hypermutation and isotype switch recombination occur in germinal center B cells, are linked to transcription, and are similarly affected by deficiency in MutS homologue (MSH)2. Class-switch recombination is abrogated by disruption of genes encoding components of the catalytic subunit of DNA-dependent protein kinase (DNA-PK(cs))/Ku complex and likely involves nonhomologous end joining (NHEJ). That somatic hypermutation might also be associated with end joining is suggested by its association with the creation of deletions, duplications, and sites accessible to terminal transferase. However, a requirement for NHEJ in the mutation process has not been demonstrated. Here we show that somatic mutation in mice deficient in NHEJ can be tested by introduction of rearranged immunoglobulin and T cell receptor transgenes: the transgene combination not only permits reconstitution of peripheral lymphoid compartments but also allows formation of germinal centers, despite the wholly monoclonal nature of the lymphocyte antigen receptors in these animals. Using this strategy, we confirm that somatic hypermutation like class-switching can occur in the absence of recombination-activating gene (RAG)1 but show that the two processes differ in that hypermutation can proceed essentially unaffected by deficiency in DNA-PK(cs) activity.
Figures
Comment in
-
Error-prone candidates vie for somatic mutation.J Exp Med. 2000 Nov 20;192(10):F27-30. doi: 10.1084/jem.192.10.f27. J Exp Med. 2000. PMID: 11085756 Free PMC article. No abstract available.
References
-
- Frey S., Bertocci B., Delbos F., Quint L., Weill J.-C., Reynaud C.-A. Mismatch repair deficiency interferes with the accumulation of mutations in chronically stimulated B cells and not with the hypermutation process. Immunity. 1998;9:127–134. - PubMed
-
- Rada C.A., Ehrenstein M.R., Neuberger M.S., Milstein C. Somatic hypermutation in MSH2 deficient mice is more focussed on intrinsic hotspots suggesting targeting to be a two stage process. Immunity. 1998;9:135–141. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
